
クオリス・イノーバが定期開催するセミナーです。
QFS00: FDA 入門コース
QFS01: FDA GMP(QSR)2日間コース
QFS02: FDA設計管理 2日間コース
QFS03: CAPAコース
QFS04: FDAプロセスバリデーションコース
QFS05: ソフトウェアバリデーションコース
QFS06: FDA QSR内部監査員コース
QFS07: FDA MDR・CARコース
最新のセミナー開催予定は、
![]()
会社で実施するオンサイトセミナーに関するお問い合わせは、
![]()
クオリス・イノーバは、医療機器・IVD に特化した FDA 規制要求事項のセミナーを開催しています。 FDA査察準備の為だけではなく、医療機器を開発・製造する日本の企業の皆様に医療機器開発の本質をお伝えし、病に苦しむ患者様のために価値ある製品を提供して頂くため、数多くの FDA 査察経験、企業内監査から得られたクオリス・イノーバだけの特別な実践的なセミナーです。
QFS00: FDA 入門コース
QFS01: FDA GMP(QSR)2日間コース
QFS02: FDA設計管理 2日間コース
QFS03: CAPAコース
QFS04: FDAプロセスバリデーションコース
QFS05: ソフトウェアバリデーションコース
QFS06: FDA QSR内部監査員コース
QFS07: FDA MDR・CARコース
最新のセミナー開催予定は、
![]()
会社で実施するオンサイトセミナーに関するお問い合わせは、
![]()

グローバルを視野に置くと米国のマーケットへの参入は必須です。企業が海外に進出する際や企業を買収する際に日本企業に最も不足しているのが、グロバールな組織体制とグローバル品質システムです。そこで、品質システムについては最も厳しい米国FDAの規制(QSR) に準拠したものづくりを行うべきです。しかしそれには、既存のISOマインドを変革した上、グロバールな感覚も身につける必要があります。
クオリス・イノーバのセミナーは、ただFDA査察にパスすることだけではなく、患者様にフォーカスした医療機器開発の本質を説くため、参加者の固定概念が一変し、新鮮な製品開発の概念が、社内の多数のスタッフのマインドを一気にポジティブに変え、会社に大きな変化をもたらすことができます。さらにグローバルヘルスケアカンパニーでの経験からグロバール企業のマインドも理解できます。
クオリス・イノーバでは、FDAの要求事項を重荷にならない、むしろ会社に変化をもたらす医療機器メーカーが知って おくべきメソッドとして紹介しています。
FDA査察準備のためのGAP監査を行う場合、必ずオンサイトセミナーを実施させていただいております。 FDA査察準備のページをご参照下さい。
![]()
オンサイトセミナーの資料請求・お問い合せはこちらから
![]()

ISOだけの品質システムを構築、運営してきた企業にとって、FDA QSR( Quality System Regulation )を理解する事とは、その差分を理解することであると一般的に思われがちです。 しかし、米国民を守るというミッションを掲げて海外までくるFDA査察と比べる事がそもそも間違っています。 ISO監査員はシステムが維持されているかと問い、FDA査察官はなぜ品質不良が発生するのかと問うのです。 この事を理解するためには、QSRを補足説明しているFDAガイダンスを理解する必要がありますが、本コースでは、FDA査察官向け査察ガイダンス(QSIT)や、FDA査察解析データを元にFDA規制要求事項の本質をわかりやすく説明します。 また、全世界に100以上の工場を持つGEヘルスケアのコーポレートオーディターとして世界の工場を監査した豊富な実績とFDA査察経験、GEシックスシグマ・ブラックベルトとして多くの品質改善経験をしてきた実績から医療機器開発の本質を鋭く説くコースです。 ( 2日コース )

セミナーに参加していただいたお客様の声です。
* QSR,設計管理ガイダンス、CAPAの3コースを受講させていただきました。 私がこれまで受講した中で最も分かり易い内容で感銘を受けました。 是非とも我が社の経営幹部に受講させたいと思います。
* まず、FDA当局の考え方とその本質を、実体験を通して説明して頂けたので、とても臨場感があり、危機感を持って聞くことができた。 また、FDAの考え方はとても論理的かつ実用的なものであり、開発効率が質を向上させるという意味に於いてもとてもよい話しを聞けたと思う。 厚生労働省とFDAの比較は、我が国の現状を明確に言い当てている。
* 査察に対応すればOKとの認識ではなく 、むしろ品質向上さらには企業価値のアップになるとの認識は大切であり、是非大事にしたいポイントです。 この点は社員全員に徹底すべきです。
* ~品質を向上させるためには、QSRへの対応が最重要であるという認識を持った。 全ての過程が根拠に基づくエビデンスで保証される。 いわゆる曖昧さを排除する仕組みと考えられる
![]()
![]()

ISOとQSRの決定的な相違点は、規制を補足説明するガイダンスをFDAが出しているということ。 そして、FDAは製品品質にフォーカスを当てると何が問題なのかを、査察やリコール、有害事象報告 ( MDR ) の結果から統計的に理解していて、ガイダンスを示すことによりQSRを補足説明しています。 さらにFDAは、品質不良の根本原因は設計不良がほとんどであると統計的に説明しており、そのため、「設計管理ガイダンス」を示して設計管理の基本的な要求事項を解説し、医療機器開発の本質を説明しています。 本コースでは、21CFR820.30(設計管理)の要求事項をFDA設計管理ガイダンス、QSIT(FDA査察官向け査察ガイド)を用い、豊富な査察経験を通してしか得ることの出来ない設計品質の本質を品質工学、タグチメソッドを交えて解説するコースです。GEシックスシグマ・ブラックベルトとして多くの品質改善の経験のあるクオリス・イノーバならではの他にはない価値あるコースです。 設計担当者のみならず、製品開発(マーケティング)、品質保証、製造担当者などボーダレスな参加者が出席することで、医療機器開発の本質に気付き、マインドがポジティブに変わり、むしろ重荷にならない、目的を持った製品開発フレームワーク構築できるようになります。 (1日コース)
セミナーに参加していただいたお客様の声です。
* 単にFDAをクリアするという目先のゴールにとらわれず敢えて高いゴールを企業文化的な視点を入れる事で目的意識を高く持つことができた。
* FDAの講習会と言うことで規格の説明が主かと思っていましたが、よりよい商品開発のための手法を聞いているようで非常に参考になり、かつ直していくべき問題点も多数ありました。
* 通常は査察に対する準備・ポイントを意識しがちになりますが、FDA設計管理ガイダンスを利用して品質向上に活かすという話しは、はじめは以外に感じましたが、納得する部分も多くありました。
* FDAが何を背景に厳しくエビデンスを求めてくるか理解できました。 うまく活用し、開発に役立てる事が本質で、FDAのための資料作りにならないように準備しようと思いました。
* 講習を受講する前は、FDA査察時のノウハウ、テクニック的な話しが中心かなと思っていましたが、実際には、製品品質向上の為にどのように設計や環境を整え、取り込むべきなのかという話しが中心で非常に有用であったと思います。
![]()
![]()

FDAの査察結果を統計的に調べてみると、FDAの査察で最も多い指摘のカテゴリーがCAPAであることをご存じでしょうか? CAPAとはISO13485 8.5改善の項目で是正処置( CA )、予防処置( PA )として規格化されているのでどの会社でもお馴染みです。クオリス・イノーバのGAP監査でも、まともにCAPAを理解して真の品質改善を実施している企業は今まで見たことがありません。その原因はISOを間違って理解している事とTQMを実施していないからです。そのため、GHTFガイダンスではその間違いを正す異例な序文から始まっています。FDAは品質を改善するためのCAPAの結果を見ることで、規制順守のみならず 、企業の品質改善活動に対する姿勢を確認しようとします。それだけではなく、このCAPAは品質改善活動してよく知られるシックスシグマのツールとよく似ており、シックスシグマをわざわざ導入することなくCAPAで品質改善ができます。 本コースでは、GEシックスシグマブラックベルトとして多くの改善経験を持つクオリス・イノーバならではの、シックスシグマの要素を交えた解説に加え、Warning Letter で指摘されている実例を元に、QSITを用いてCAPAをわかりやすく解説し、コース終了時には、適切なCAPAのフォーマットを完成させ、どう運用すればシックスシグマ活動のような品質改善結果を得られるのかを理解し、企業で実践することができるようになります。
ミナーに参加していただいたお客様の声です。* 十数年間ISO品質システムの体制下で物事を判断してきましたが、なかなか品質改善に結びつかないことが多くありました。 しかし、CAPAでの考え方を入れることで品質改善の可能性が見えてきました。
* GEの実例を基に説明してもらい、CAPAの基本的な考え方がわかりました。 実際に運用していくことがポイントだと思います。
* 是正処置と予防処置が両方実施されることはない。 これは目からうろこでした。
* 是正と予防は全く別の運用をしなければならないことがよくわかりました。 また、(CAPAのフォーマットについて)自社の内容に不足が多くあることがわかり大変参考になりました、
* FDA対応として取り組むよりは、品質改善として取り組むと言う考えに共鳴しました。
![]()
![]()

工程管理はどのパラメータで監視していますか?この質問から始まる工程査察は、工程設計のエビデンス、工程管理パラメータの科学的な根拠の無さを次々と露呈させていきます。工程バリデーションの主たる目的は、試験に頼らないパラメータ監視で実現する中間製品の品質確保にあります。FDAが求めているのは、試験や検査に頼らないパラメータ監視によるバラツキの防止にあります。FDAは工程設計も 820.30 に相当する設計行為と明言しており、設計移管、即ち工程設計とプロセスバリデーションは同時に発生するものです。このコースでは、FDA、GHTFの各ガイドラインを元に、ほとんど理解されていない工程検証、工程バリデーションの違い、設備バリデーションと工程バリデーションを行う為のプロセス( VMP,DQ、IQ、OQ、PQ、PPQ )を品質工学、タグチメソッドを用いた手法で紹介しするコースです。
セミナーに参加していただいたお客様の声です。
* プロセスバリデーションについて、概要は理解していたつもりでしたが、全体的に説明を受け新たに気付いた点も多くあった。 DQの考え方、QC工程表を利用したバリデーションの決定、リスクベースの考え方が参考になりました。
* 参考書等で、DQ,IQ、OQ,PQの用語が出てきて、内容を読んでも理解しづらかったのですが、それが理解できました。
* 現在、査察に向け準備しているものがあるが、セミナーを受講し、確認すべき点が明確になった。 特にリスクベースドバリデーションはの考え方やCAPAシステムとの繋がりは考えるべきと感じた。
* 今回のセミナーは、実務的な内容があり、普段疑問に思っていたことも回答してもらい、非常に有意義であった。
![]()
![]()
過去のセミナー情報です。
タイトルをクリックすると本文がご覧頂けます。
購買プロセス リモートコース
〜 FDA 査察指摘 Worst 4 購買プロセスの本質と対策 〜
21CFR Part 820・ISO13485・ISO9001・ガイダンス
このコースの売り上げ金の一部は人道支援活動に寄付されます
リモートで開催され会場開催はありません
☆ 開催概要 ☆
FDA査察で必ず指摘を受ける(ワースト4)重要なプロセス。アセンブラーが多くなった昨今、行政当局も購買プロセスに多くの査察時間を費やすようになりました。また、重要構成品のアウトソースを行う企業がほとんどで、実際はアウトソースとして扱うべきところを、パーツサプライヤとして扱ってみたり、マネジメントシステムの傘下として扱うべきサービス購買の要求事項を理解していません。さらに、購買プロセスは、言われた通りにモノを買っていればよいというものではなく、サプライチェーン全体を俯瞰し、製品企画時の上流から積極的にコンカレントで参画、最も効率よいQCDを提案することこそ会社に貢献するプロセスとして価値があります。その為には、戦略的な組織体制とフレームを構築する必要があり、開発購買や情報購買と言ったフレームワークは早期の製品ローンチを実現します。新たな規制QMSRを想定し、ISO13485、9001の要求も加味しながら理解を深めます。
オンサイト(御社のみで)でも可能です。お問い合わせはこちらから。
日 時:2025年6月16日(月曜) 9:00 〜 16:30
会 場:リモートのみ
( リモートは ZOOM でのご参加となります )
講 師:クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象: 医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
ODM、OEMとして設計・製造を受託している企業
受講料: ¥55.000-( 消費税込、テキスト事前郵送 )
割 引: ¥44.000-( 消費税込、テキスト事前郵送 )
1社2名様以上ご参加の場合
特 割: 弊社プロジェクト実施中、継続教育等の場合ご相談下さい。
定 員: リモート 定員 40名
( 出席者全員に教育記録として利用できる修了書をお渡しします )
申込締切: 6月9日(月曜)
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
![]()
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
A.FDA規制、査察の最新情報と傾向分析(QMSRの動向)
1.適用規制・規格
購買プロセスへの規制
ガイダンスから学ぶ
2.プロセスアプローチの理解
重要構成プロセスであることの理解
プロセスの役割
3.購買管理の目的
購買プロセスの目的
製品購買とサービス購買
購買製品の検証(受入検査)
4.用語の定義
製品購買、サービス購買とは
プロセスのアウトソースと部品購買の相違
アウトソース先の管理、監視
5.購買管理
GHTF( IMDRF )ガイダンスに基づく手順
購買管理の7ステップ
6.購買方針
購買方針のない購買は不正を生む
7.戦略・開発購買
買プロセスの製品企画への関わり方
戦略購買
開発購買
情報購買
開発スピードを上げ原価削減、購買活動のスピードを上げある
ベストプラクティスの収集と技術の取り込み
8.購買計画
製品企画段階から参画
供給する製品、サービスの特定
技術及びプロセス情報の特定
供給者候補の特定
リスク、管理方法の特定
9.供給者候補選定
供給者ビジネス、運用能力評価
供給者候補選定
10.供給者選定
評価選定基準の策定
供給業者候補とのコミュニケーション
供給者候補の能力評価
供給者の承認
11.管理方法の取り決め
購買情報とは
管理方法
購買製品の検証
12.変更時対応
変更管理規定
契約変更通知
4Mと変化点管理
13.納期、測定、監視
購買製品の監視QCD
リスクベースに応じた管理、監視
SQCを用いた分析、改善
金型管理
14.フィードバック、コミュニケーション
CR
SCAR
供給者評価シート
15.サプライヤー監査
リスクベース監査
5つの監査、評価エリア
情報購買、戦略上重要な監査チーム
16.物流(ロジスティクス)
取扱、倉庫、輸送管理
( 内容が変更になる場合もございますが大きな変更はございません。)

FDA 設計管理2日間HBコース
〜 安全性と性能を証明するフレームワーク 〜
NEW QMSR 対応 21CFR Part 820.30 / 75 ISO13485
QSR内部監査員コースご出席要件コースです
( オンサイト及び過去弊社コースを受講された方も要件を満たします )
会場参加、リモート、ハイブリッド形式で開催します
☆ 開催概要 ☆
ほとんどの日本企業に於ける医療機器開発・設計管理の仕組みは世界標準にほど遠く、これはグローバルに進出しようとする企業のみならず、患者の命を預かる全ての医療機器メーカーにとって大きなリスクを伴います。米国FDAは自らの査察で、医療機器品質不良の根本原因は上流側の設計管理にあると査察の結果から統計的に見抜いており、それはクオリス・イノーバの20年以上にわたるGAP監査でも明らかです。そこで本コースでは、グローバルな開発、設計管理を米国FDA「設計管理ガイダンス」及び欧米で取り入れられている開発フレームワークを元に体系的に解説、また、品質不良を発生させない仕組み(ロバスト設計)をタグチメソッドとライフサイクルマネジメントを用いて解説します。 2日目はプロセスバリデーションの本質、工程内試験に頼らない均一で高品質な製品を一貫して製造する為、パラメータによる製造方法を実現する工程設計について解説します。 ”規制” だからではなく、設計品質を高めることにより患者さんの命を守り、企業にとっても大きな潜在的利益を得られるという大原則は、エンジニアの方々から多くの共感と支持を頂いてきたクオリス・イノーバの最も人気のある価値あるコースです。 このコースはオンサイトでも実施しております。お問い合わせはこちらから。
日 時: 2025年6月23日(月曜)、24日(火曜) 9:00〜16:30
締 切: 2025年6月16日(月曜)
会 場: 全国町村会館 大ホール(東京・永田町) 永田町駅から徒歩1分( 3番出口 )
リモートで参加はZOOMを使用
講 師: クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象: 医療機器・IVD市場にこれから参入しようとしている企業
医療機器・IVDを海外に輸出しようとしている企業
FDA査察準備、MDSAPを受審しようと準備している企業
既にFDA査察を終え、指摘対応に苦慮している企業
製品実現に関わる各組織( マーケティング、開発、生産技術、品証など )
受講料: ¥99,000( 税込、テキスト・QSR訳本第5版 昼食含む )
割 引: ¥88,000( 税込、テキスト・QSR訳本第5版 昼食含む )
1社2名以上受講される場合
特 割: 再受講の方、プロジェクト終了企業様で継続研修としてご検討の方ご相談下さい。
定 員: クラス 定員 20名
リモート定員 30名
出席者全員に教育記録として利用できる修了書をお渡しします。
配 付: FDA QSR 対訳+ポイント解説(第5版)旧版を配付します。
(第6版)及び FDA QMSR 対訳+ポイント解説(第1版)は、
(5月開催)FDA QMSR/QSR/13485 コースでお渡ししています。
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡いたします。
2名様以上ご参加の場合、お名前とローマ字読み、メールアドレスを連絡欄にご記入下さい。
持参品: PC( WiFi 使用可能 )
ご注意: 本コースは、品質マネジメントシステムの一部を解説するため、システム全体の知識がないと理解できません。本コース受講前に5月開催QMSR2日間コースの受講をお勧めします。
開催要件:
リモートご参加の場合
ZOOM以外での対応はしておりません
会社からご参加の場合、会議室等お一人になれる場所から会社PCでのご参加となります。
自宅からご参加の場合、会社PCでのご参加となり、
その他、チェックリストでご確認頂く為、事前にご確認をお願いしています。
メールでお問い合わせ
電話でお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1日目(セミナー内容)
A.コンプライアンスとインテグリティー
設計不良の原因は何か
設計CAPAとは
ケーススタディ
不正を排除する仕組み
規制・規格に満足するればOKか?
人の命
行政によるミッションの相違
B.医療機器規制を理解する
新しい規則QMSRと今後の対応値
適用規制、規格、ガイダンス
C.製品実現 ISO13485 - 7
製品実現の為のプロセス構築とは
安全性と有効性の立証
ライフサイクルマネジメント(LCM)
顧客のフィードバック&コミュニケーション
1.Subpart C 設計管理 820.30 ISO13485 - 7.3
設計管理の目的とその重要性
ウオーターフォールモデル Waterfall Model
関連する ISO13485 の項目(プロセス)
Form 483 の FY 統計的解析から理解すること
2.820.30 (a) 総則 ISO13485 - 7.3.1
プロセスアプローチ ISO13485 4.1 の理解と
経営者の責任(リソースの提供とKPI)
製品実現プロセス
タートルチャート
リソースとしての力量(教育と訓練) 820.25
文書体系から俯瞰する設計管理プロセス
3.820.30 (b) 設計及び開発計画 ISO13485 - 7.3.2
医療機器開発ライフサイクルマネジメント(LCM)
設計品質確立の重要な要素; コンカレントエンジニアリング(CE)
PM、PLの役割と責任
ライフサイクルリスクマネジメントの理解(LCRM) ISO14971
タグチメソッド開発フレームワークによるフロントローディング手法
マーケティング戦略の重要性
開発計画書
4.820.30 (c) 設計インプット ISO13485 - 7.3.3
コミュニケーションエラー
曖昧なニーズの明確化
要求仕様から完成品までの一貫性
トレーサビリティーマトリックス(TMX)
ラベリング、包装設計のインプット
820.120 機器のラベリング
820.130 機器の包装
5.820.30 (d) 設計アウトプット ISO13485 - 7.3.4
タートルチャートのアウトプット
設計アウトプットのリスト
E−BOMの役割
製造仕様書とは
6.820.30 (e) 設計レビュー ISO13485 - 7.3.5
問題の早期発見、解決が目的でセレモニーで終わらせない仕組み
設計品質確立の重要な要素
設計レビューとテクニカルレビュー
レビューワーと責任と権限
7.820.30 (f) 設計検証 ISO13485 - 7.3.6
インプットからアウトプットまでのトレーサビリティー
製品の安全性、性能を証明するエビデンスの作り方
設計検証計画に必要な要素
設計検証レポートはデータの信頼性を確保
8.820.30 (g) 設計バリデーション ISO13485 - 7.3.7
設計検証と設計バリデーションの相違
設計バリデーションの手法
2つのバリデーションの相違と記録
ソフトウェアのバリデーションとVモデル
3種類のバリデーションの相違
9.820.30 (h) 設計移管 ISO13485 - 7.3.8 → 第2日目(工程設計)
10.820.30 (i ) 設計変更 ISO13485 - 7.3.9
変更管理プロセスに伴う設計変更
ライフサイクルリスクマネジメント
タートルチャート
設計変更フロー
11.820.30 (j ) DHF ISO13485 - 7.3.10 設計・開発ファイル
DHF / DMR / DHR の定義
識別、ファイルの基本
文書体系と設計標準
2日目(セミナー内容)
設計移管と工程設計
820.30 (h)、13485 - 7.3.8、7.5.6
1.820.30 (h) 設計移管 ISO13485 - 7.3.8
ライフサイクルリスクマネジメントにおける設計移管(工程設計)
均一な製品を一貫して生産する手法
プロセスバリデーションの目的
2.なぜ工程バリデーションを実施するのか 820.75, ISO13485 - 7.5.6
プロセスバリデーションの目的とその本質
プロセスバリデーションの効果
工程管理、監視の目的と本質
医療機器規制・規格の理解
3.バリデーションすべき工程とは
バリデーションすべき工程の識別
検証工程とバリデーション工程の相違
設備バリデーションと工程バリデーション
バリデーション工程例
4.バリデーションの種類
バリデーションの種類
設備バリデーションと劣化パラメータ 820.70(g), 72(a)
コンピューターシステムバリデーション(CSV) 820.70(i), ISO13485 - 7.5.6 (g)
5.バリデーションステップ 820.75, ISO13485 - 7.5.6
VMP:バリデーションマスタープラン
DQ: 設計時適格性評価
IQ: 据付時適格性評価
OQ: 稼働時適格性評価
PQ: 製造時適格性評価
PPQ: プロセス街道性評価
バリデーション文書化
プロセスバリデーションの明確化
自動化工程の明確化
CSVカテゴリーの明確化
ソフトウェアのバリデーションの基本
リスクベースCSV
6.工程設計とプロセスバリデーション 820.30 (h)、 ISO13485 - 7.3.8
ライフサイクルマネジメントに於ける工程設計
工程設計の進め方
工程設計とプロセスリスク分析( PFMEA )
環境管理 820.70(c)、 ISO13485 - 6.4
汚染管理 820.70(e)、 ISO13485 - 6.4
副資材 820.70(h)、 ISO13485 - 7.5.2
工程設計PTMX
QC工程表
7.回顧的バリデーション
8.バリデーションのアウトプット
QC工程表と統計的手法(工程能力)
リバリデーション設定
設備インスペクションとメンテナンス
( 内容が変更される場合がございます )

FDA QMSR/QSR/13485
2日間ハイブリッドコース
〜 医療機器・IVDに求められるMSの本質を学ぶ 〜
21CFR Part 820 / ISO13485 / ( ISO9001 )
このコースは会場、リモートのハイブリッド開催です。
2026年2月に施行される QSR に変わる新たな規則 QMSR について、
ISO13485 との相違はもとより、原点に立ち返る MS の本質について学ぶ新たなコースです。
年末に実施する監査員コースの受講要件となります。
☆ 開催概要 ☆
QSR翻訳本(第5版)+QMSR翻訳本(第1版)付き
『 医療機器を作ることと、安全性と性能を証明することは異なる。』このことに経営陣が気付かない医療機器メーカーが、規模の大小に関わらず法違反や患者の命に関わる事件を起こしています。 規制だけ満足していればよいという考え方が、どれだけこのビジネスでは会社のみならず社会にリスクがあるかのを最初に理解すべきです。
他方、市場を考えると、米国が最も大きなマーケットであることは医療機器でも例外では無く、米国での成功がグローバルを制すると言っても過言ではありません。しかし、米国で製品を上市するには医療機器規制 QMSR に基づく MS を構築し、『安全性と性能』を証明しなければなりません。2026年2月から新たに適用となるこの規則は、FDA が QSR を計画的に反映させた ISO13485(2016) が基本となります。今後は QSR の欠点でもあった MS の本質、リスクベースプロセスアプローチのフレームワークが必須となります。弊社の経験上 ISO1345 取得済みの企業であっても ISO に準拠しているとはとても言いがたく、FDA の査察アプローチは ISO と異なる為、 QMSR には準拠できません。今まで弊社は一貫して規制の本質を解き、事業活動とのインテグレーションを取ることにより、結果の出せるマネジメントシステムを提唱し、多くの企業の皆様の共感を集め支持されて参りました。米国に進出する企業のみならず、医療機器・IVDを開発しようとしている企業の皆様のマインドが大きく変わる価値あるクオリス・イノーバのメインコースです。
新規制が適用となるまで QSR が適用となる為、本コースでは現行 QSR, QSMR, ISO13485 について、リスクベースプロセスアプローチ、LCMといったシステムの根幹となるフレームワークを中心に概要を説明します。
オンサイト(一定人数が集まった会社でのセミナー)も可能です。お問合せはこちらから。
日 時: 2025年5月26日(月曜)、27日(火曜) 9:00〜16:30
締 切: 5月19日(月曜)
会 場: 全国町村会館(東京・永田町)(永田町駅から徒歩1分)
リモートで参加なさる場合、ZOOMでの御参加となります。
講 師: クオリス・イノーバ株式会社
代表取締役社長 木村 浩実
対 象: 医療機器・IVD市場にこれから参入しようと考えている企業
米国に輸出を計画している企業
米国FDA 査察準備を考えている企業
ODM、OEM として医療機器を受託しようとしている企業
受講料: ¥99,000−(税込)( 2日分、テキスト、対訳本2冊、昼食代含 )
リモートで参加なさる方には事前にテキスト、対訳本を郵送いたします。
昼食は会場ご出席者のみ
割 引: ¥88,000−(税込)
1社2名以上ご参加の場合
特 割: 企業で行うQMS構築プロジェクトなどや企業で弊社が実施した継続研修などで
ご利用の場合、特別割引を適用させて頂きますのでメールでご相談下さい。
満席となった場合、正規料金をお支払いの方が優先となりご希望に添えない場合
がございますので予めご了承ください。
定 員: クラス 定員 20名様
リモート定員 30名様
( 出席者全員に教育記録として利用できる修了書をお渡しします )
開催要件:
1.リモート参加要件
会社からの場合、個室(会議室等)から会社PCで御参加となります。
ご自宅からの場合、会社PCでのご参加となります。
その他チェックリストを事前にお渡しし環境をご確認頂きます。
2.うぃるす対策
弊社のウィルス対策を順守して頂きます。
当日発熱がある場合、参加を御控え頂きます。
お申込: セミナー申込みページからお申し込み下さい。
折り返し、会場、お振り込み先情報などをメールでご案内致します。
2名以上で参加なさる方ははその旨ご記入下さい。
割引料金でご案内致します。
![]() メールでのお問い合わせ
メールでのお問い合わせ
![]() 電話でのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.医療機器を作ることと、安全性、性能を証明することは異なる
なぜ承認が取得できないのか
なぜ査察で警告を受け裁判にまで発展するのか
2.コンプライアンスとインテグリティー
患者の命と会社の危機管理
データインテグリティー
3.医療機器規制の理解
行政によるミッションの相違
米国規制法
医療機器規制の理解
QSR vs QMSR
4.FDA査察
FDA査察の最新動向とISO企業の実態
FDA査察、頻度、査察タイプ
QSITアプローチ
A リスクベースプロセスアプローチ
Subpart_B 品質システム要求事項
820.20 経営者の責任
プロセスアプローチ
タートルチャート
マネジメントレビュー
文書体系
品質監査
力量
Subpart_D 文書管理
820.40 文書管理
Subpart_O 統計的手法
820.250 統計的手法
Subpart_J 是正及び予防処置
820.100 CAPA
用語の正しい理解
エスカレーション
プロセスアプローチ
B 製品実現フレームワーク
Subpart_C 設計管理
Subpart_E 購買管理
Subpart_F 識別及びトレーサビリティー
Subpart_G 生産及び工程管理
Subpart_H アクセプランスアクティビティー
Subpart_I 不適合製品
Subpart_K ラベリング及び包装管理
Subpart_L 取扱い、保管、流通及び据付け
Subpart_M 記録
Subpart_N 附帯サービス

〜 FDA査察指摘ワースト&キープロセスを理解する 〜
NEW QMSR 対応 21CFR Part 820.100、ISO13485 8.5、ISO9001 10
このコースの売上金の一部は人道支援に寄付されます
* コース概要 *
CAPAプロセスを確認するだけで、その会社の品質システムや品質に対する理解度がよくわかります。CAPAプロセスに問題があれば、品質マネジメントシステムの適切性、妥当性はなく、特にマネジメントレビューがまともに運用されているとは思われません。品質マネジメントシステムの原則はプロセスアプローチ、PDCAをまわす為、マネジメントレビューのインプットでプロセスを監視し、改善ツールであるCAPAでプロセスを改善します。当たり前のことがシステムとして運用されていないことを理解している規制当局は、CAPAプロセスにフォーカスして確認しようとします。QMSが機能していないことは、昨今の品質に関わる事件から明らかで、医療機器・IVDの場合、命に関わる大問題です。本コースでは今までの内容を大きく変更し、プロセスアプローチの手法に重点を置き、事例を元にCAPAの本質を理解して頂きます。 このコースは受講者と同じ目線の御社事例を用いることでより理解が高まるオンサイトセミナーが可能です。お問合せはこちらから。
日 時:2025年3月10日(月曜)に延期 9:30 〜 16:30
締 切:2025年3月4日(火曜)AM
会 場:リモートのみ
( 参加はZOOMのみとなります )
講 師:クオリス・イノーバ株式会社代表取締役社長 木村 浩実
対 象:医療機器、IVD、医薬品等に限定せず、CAPAプロセスを学びたい企業様
受講料: ¥55.000-( 税込、テキスト代 )
事前にテキストを郵送致します
弊社FDAQSR第6版を特別価格 ¥1,650−(税込)で希望者に販売
割 引: ¥44.000-( 税込、テキスト代 )
1社2名様以上ご参加の場合
特 割: 弊社プロジェクト実施中、継続教育等の場合ご相談下さい
定 員: リモート定員 60名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
開催要件:
1.リモート参加要件
会社からの場合、個室(会議室等)から会社PCで御参加となります。
ご自宅からの場合、会社PCでのご参加となります。
その他チェックリストを事前にお渡しし環境をご確認頂きます。
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
![]()
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.FDAがCAPAに注目している理由
警告書の傾向分析
2.なぜ品質問題は再発するのか
事例から理解する
直接原因と根本原因の相違
再発品質不良の根本原因
3.CAPAを理解する本質
リスクベースプロセスアプローチとは?
9001,13485CAPA
測定、分析、改善
CAPAはプロセス改善ツール
4.TQMの仕組みが不可欠
トップがマネジメントするシステム(MS)
プロセスアプローチのPDCAを廻す
プロセスKPIとは
ガイダンスが示すCAPAプロセス
5.リスクベースプロセスアプローチ
9001,13485CAPA
リスクベースプロセスアプローチとは?
測定、分析、改善
6.規制・規格の理解
CAPAの規制、ガイダンス
是正、予防、水平展開の意味を理解する
統計的手法を使用し最初に予防
予防するマネジメントシステムが重要
7.用語を正しく理解する
修正、是正、予防の相違
逸脱、不適合、逸脱許可と特別採用の相違
8.ガイダンスに基づくCAPA手順
Phase 1 データソースKPI
Phase 2 データ分析
Phase 3 CAPA
Phase 4 マネジメントレビュー
9.CAPAプロセスエスカレーション
CA・PAエスカレーションの仕組み
リスク判断基準
10.フィードバック
フィードバックプロセス
CAPAはシステム改善プロセス
システムの有効性
11.CAPA7ステップ
Step 1 問題を検出し明確にする
Step 2 問題の原因を詳しく調査する
Step 3 直接、根本原因の特定
Step 4 処置の特定と検証または妥当性確認
Step 5 予防または是正する為の処置
Step 6 処置の結果を文書化
Step 7 有効性の確認
12.品質問題の事例に学ぶ
Warning Letter Case Study
実際の事例

FDA MDR&CAR リモートコース
リモート形式
〜 査察指摘リスクが高い行政報告プロセス 〜
21CFR : Part 820, 803, 806, ISO13485
このコースの売り上げ金の一部は人道支援活動に寄付されます
☆ 開催概要 ☆
新しいQSRに変わるQMSRでISO13485にないプロセスを学び早期にプロセスを構築する必要があります。FDA査察レポートの解析からワースト5位(全体の約10%)に急浮上しているMDR(有害事象報告)、日本でも Warning Letter (警告書)を受けているほとんどの企業がMDRの指摘を受けています。昨今のデータ偽装事件を受け、FDAはこれらにフォーカスし厳しく査察を行っています。医療機器を作っているメーカーは、患者さんの命を守るという社会的な責任があります。会社を守っているようでも、結果的に会社が倒産に追い込まれることもある非常にリスクの高いプロセスであることを理解すべきです。それには規制の正しい理解、規制を守る仕組みと組織が必要です。このセミナーでは、CAPAサブシステム(苦情、不適合、CAPA)の各プロセスとリンクし、日本のメーカーの Warning Letter (警告書)を引き合いに正しいMDRプロセスと関連するCAR(リコール)プロセスを構築することを理解します。
このコースはオンサイトでも実施しています。お問い合わせはこちらから。
日 時:2025年3月17日(月曜)9:30 〜 16:30
3月11日(火曜)締切
会 場:リモート(ZOOM)のみ
( ZOOMはブラズザからログインできます )
対 象:既に医療機器・IVDを米国に輸出している企業
これから医療機器・IVDを米国輸出する準備を行っている企業
既にFDA査察を終え対応に困っている企業
受講料: ¥55,000-( 消費税込み、テキスト代含む )
リモートの方には事前にテキストを郵送させて頂きます
割 引: ¥44,000-( 消費税込み、テキスト代含む )
1社2名以上ご出席の場合
定 員: リモート(30名)
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
特 割:弊社プロジェクトを実施中、又は継続研修をご希望の場合、負担軽減のためさらに
割引をさせて頂きますのでお問い合わせください。この場合、正規料金でお申込の方
が優先され、出席できない場合がございますので予めご了承ください。
お申込:セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
開催要件:リモート参加の場合、リモート環境に確認とNDAへのサインをお願いしています。
折り返し、リモートリンクお振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.規制を理解する
過去事例と統計からみる規制違反
理解すべき規制・規格・ガイダンス
eMDR規制
苦情組織と力量
規制が求めるあるべき組織と力量
プロセス構成
2.有害事象報告( MDR )Part 803
MDR規制の基本要件
FDA Warning letter から
規制の目的、重要性
MDRとして報告すべき事象とは
2.1 定義を理解する
重篤な障害
機能不全
報告すべき Malfunction
合理的に推測する
認識する
2.2 4.5日、30日報告
報告形態と時間軸
5日報告
ディシジョンツリー
2.3 FDAへの報告
報告フロー
報告様式 3500A
報告不要の場合
報告書の種類
報告免除
Warning letter 警告書 !
2.4 MDRプロセス
MDR報告フロー
CA,PAプロセス
3.回収報告(CAR)Part 806
規制の理解
報告免除
定義
リコールフロー
任意と強制リコール
定期メンテナンスとリコール
3.1 リコール判断基準
リコール判断基準例
ラベリングの定義
ラベリング違反と法的根拠
報告すべきラベリング違反例
3.2 CAR ディシジョンツリー
リコールディシジョンツリー
3.3 報告書と処理フロー
リコール判断後フロー
リコールクラス
有効性の確認
CAPAとの関係
4.警告書から理解すべき注意点とプロセスへの展開
Warning Letter ( 警告書 )の事例から理解する
警告書から理解すべき注意点とプロセスへの展開
プロセス構築後のレトロスペクティブな是正とは
*コース内容が一部変更される場合がございますが大きな変更はございません。

FDA QMSR マネジメントシステム1日コース
〜 結果を出すマネジメントシステムの基礎を学ぶ 〜
FDA QMSR / ISO13485
このコースの売上金の一部は人道支援活動に寄付されます
☆ 開催概要 ☆
ISO認証を取っているからと米国FDA査察を軽く見た結果、FDAから警告( Warning letter )を受け、刑事裁判にまで発展し、株価は下落、裁判費用や改善コストに多額に費用が掛かってしまった。規制や規格の目的や意味も教えないで仕事をさせた結果、人の命に関わるような事件にまで発展させてしまう。規制だけ満たしておけばいい、余計なことをするなと上司から指示され、結果、重大なリコールを発生させてしまう。これが日本の悲しい現実。根本原因は、規制、規格の教育、継続訓練がプロセスとしてなく、経営者やマネージャーも規制の本質を理解していない。そのため、いきなり弊社のFDAQSR2日間コースを受講してもさっぱり理解できないのが現実。まず、基本からと弊社も日本の現状に気付き、忙しい方々に1日でマネジメントシステムの基礎を理解して頂くリモートコースです。
オンサイトでも可能です(お問合せはこちらから)
日 時: 2025年2月3日(月曜)開催 9:30〜16:30
締 切: 2025年1月28日(火曜)
会 場: リモート (ZOOMのみ)
講 師: クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
受講料: ¥55,000( 税込み、テキスト、QSR、QMSR飜訳本付き )
割引き: ¥44,000( 税込み )1社2名以上ご参加
特 割: 弊社MS再構築プロジェクトや継続研修を実施しておられるクライアント様はご相談ください
企業でまとまった人数で受講なさる場合もご相談戴けます
定 員: 50名
対象者: 1.経営者、マネジメントチーム
2.QMSRの概要を理解したい方
3.直接もの造りに関与しないプロセス(人事、総務、営業など)
4.1日でマネジメントシステムの概要を理解したい方
出席者全員に修了書を発行します
お申込: セミナーお申込ページよりお申し込み下さい
![]()
メールでお問い合わせ
電話でお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.医療機器を作ることと、安全性、性能を証明することは異なる
− なぜ承認が取得できなかった?
− なぜ査察で警告を受けてしまった?
− 間違えてはいけない企業の優先順位
− 警告書を見れば企業の姿勢がよくわかる
2.結果を出すためのQMS
− プロセスをマネジメントするプロセスアプローチとは
− マネジメントのツールであり、ビジネスの結果を出すツール
3.MSの原則、プロセスアプローチを理解する
− リスクベースプロセスアプローチ(RBPA)とは
− プロセスを管理するタートルチャート(TC)
− コンピテンス(力量)を付与して働いて貰う
− KPIを監視し予防する(PA)
− 品質マネジメントシステム計画
4.PDCAで予防するフレームワーク
− マネジメントレビューでKPI監視
− プロセス改善ツール(CAPA)
− マネジメントレビューで監視、指示
5.データを分析しフィードバックして予防する
− データソース(規制、ガイダンスで決められている)
− データ分析と予防処置
− TQA会議とQAの役割
6.プロセスを改善する為のCAPA
− 問題を起こさないための予防活動(是正では手遅れ)
− CAPAプロセスを見ればその会社のシステム理解度がわかる
− エスカレーションCAPA
− プロセスオーナーの責任と役割
− プロセスアプローチの改善ツール
7.SCMを俯瞰した購買プロセスとは
− アウトソースプロセスの説明責任者は?
− 戦略購買と開発購買
− 受入検査プロセスは購買プロセス
8.システム構成と文書管理
− 手順書の作り方を見れば、システムをまともに運用できているかどうかが判る
− システム構成(QM、STD、SOP、WI)
− システム体系図(ツリー図)
− 文書作成の基本(ワークフローに合わせた手順書作りとは)
9.規制当局が求めるライフサイクルマネジメント(LCM)
− コンプライアンスを堅持、説明責任を果たす
− コンカレントエンジニアリング(CE)の役割
− ライフサイクルリスクマネジメント(LRCM)が必須( 14971 )
10.安全性、性能の説明責任を果たすフレームワーク
− ウオーターフォールモデル(WFM)
− ロバスト設計
− トレーサビリティーマトリックス(TMX)
− プロセスバリデーション(PV)
11.説明責任を果たすデータインテグリティー
− データが真実であることを証明する手法
− デジタルシステムバリデーションとは

FDA QSR/QMSR 監査員コース Rev.17
〜 QSR 対応内部監査員養成のための社内力量認定2日間教育コース 〜
* FDA QSIT をベースにプロセスアプローチで実施することを理解します *
このコースの売り上げ金の一部は人道支援活動に寄付されます
☆ 開催概要 ☆
FDAの監査資料の結果から、品質監査( Part 820.22 )の指摘は、もうすぐワースト10に入るほど急上昇しているセクションです。FDAは、いい加減な監査報告が経営者に報告されていることが根本原因であることに気付いています。プロセスアプローチでは、マネジメントシステムは経営ツールでもある為、組織ではなくプロセスという切り口で監査を行い、適切性、妥当性を判断し、経営者に報告、プロセスオーナーが改善を廻すというPDCAの枠組みが必要です。一方、現実はプロセスを監視し傾向管理を行っておらず、訓練もされていないにわか監査員が片手間に監査を行っているだけ。これは医療機器ビジネスでは致命的で、リコールで何十億の損失を出すだけではなく、人の命に関わる重大な問題に発展し、会社の存続まで危ぶまれます。このコースでは、ISO内部監査員の資格を持つ方々がQSRの内部監査が行えるよう社内資格を付与するための社内資格認定コースとして位置づけられます。一般に行われているISOの内部監査手法とは異なる、付加価値を生む予防対策としての監査手法とノウハウを学びます。( このコースを受講した後、オンサイトでクオリス・イノーバによる内部監査のOJTを行うとより効果的です )
日 時:2024年12月2日(月曜)、3日(火曜) 9:30 〜 16:30
締 切:2024年11月26日(午前中)
会 場: 全国町村会館(東京・永田町) 永田町駅から徒歩1分( 3番出口 )
対 象:すでに弊社主催下記コースをオープン、オンサイト問わず受講された方限定です。
* FDA GMP QSR 2日間コース
* FDA 設計管理2日間コース
このコースはQSRの基礎知識が無いと受講できず、弊社のコースを受講されていない場合
異なる解釈により内容をよく理解出来ません。 何卒ご理解をお願い申し上げます。
受講料: ¥99,000( 税込、テキスト・QSR飜訳本第6版、昼食含む )
割 引: ¥88,000( 税込、テキスト・QSR飜訳本第6版、昼食含む )
1社2名様以上受講される場合
定 員: 20名限定 満席になり次第締切ます
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
過去に受講された弊社QSR、設計管理各コースの受講日を記載して下さい。
オンサイトで受講された場合でも構いません。
折り返し、会場、お振込先情報をメールでご連絡致します。
セミナーに参加するための宿題を追って発送し致します。
2名様以上ご参加の場合、お名前とローマ字読み、
メールアドレスを連絡欄にご記入下さい。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.内部監査の目的を理解する
規制・規格が求めていること
患者の命を優先する監査とは
企業の付加価値を高める為には
2.内部監査員に求められる力量
内部監査員とリーダーに求められる力量
内部監査員訓練
マネジメントチームのカウンセラー
3.プロセスアプローチ
CAPAの理解とCAPAアプローチ
プロセスのR&R( 役割と責任 )を明確にする
プロセスを常に測定し改善する
内部監査はプロセスに注目
4.監査判定基準
チェックリストと数値化
プロセス毎に数値化
FDA査察と内部監査判断基準の相違を知る
5.監査を計画する
マネジメントレビューと品質会議からの指示
リスクベース&プロセスアプローチによる監査計画
プロセス管理パラメートと傾向分析結果
是正監査と予防監査の相違
監査は短期集中型で実施
監査員レビュー会のノウハウ
監査報告書記載のポイント
6.内部監査レポート
トップマネジメントに対する報告
プロセスの不足、改善点を報告
規制に対するコンプライアンス状況の報告
トップマネジメントのアクション
7.監査の目のつけどころ( QSIT )
QSITに基づく監査手法と勘所
プロセスアプローチの手法を全員が理解
1.経営者による管理
2.設計管理
3.CAPA
MDR報告
CAR
MDトラッキング
4.P&PC
5.滅菌工程
+6.各プロセス
*内容は変更される場合がございます*

〜 FDA査察指摘ワースト&キープロセスを理解する 〜
NEW QMSR 対応 21CFR Part 820.100、ISO13485 8.5、ISO9001 10
このコースの売上金の一部は人道支援に寄付されます
* コース概要 *
CAPAプロセスを確認するだけで、その会社の品質システムや品質に対する理解度がよくわかります。CAPAプロセスに問題があれば、品質マネジメントシステムの適切性、妥当性はなく、特にマネジメントレビューがまともに運用されているとは思われません。品質マネジメントシステムの原則はプロセスアプローチ、PDCAをまわす為、マネジメントレビューのインプットでプロセスを監視し、改善ツールであるCAPAでプロセスを改善します。当たり前のことがシステムとして運用されていないことを理解している規制当局は、CAPAプロセスにフォーカスして確認しようとします。QMSが機能していないことは昨今の品質に関わる事件から明らかで、医療機器・IVDの場合、命に関わる大問題です。本コースでは今までの内容を大きく変更し、プロセスアプローチの手法に重点を置き、事例を元にCAPAの本質を理解して頂きます。 このコースは受講者と同じ目線の御社事例を用いることでより理解が高まるオンサイトセミナーが可能です。お問合せはこちらから。
日 時:2024年11月8日(金曜)9:30 〜 16:30
会 場:リモートのみ
( 参加はZOOMのみとなります )
講 師:クオリス・イノーバ株式会社代表取締役社長 木村 浩実
対 象:医療機器、IVD、医薬品等に限定せず、CAPAプロセスを学びたい企業様
受講料: ¥55.000-( 税込、テキスト代 )
事前にテキストを郵送致します
割 引: ¥44.000-( 税込、テキスト代 )
1社2名様以上ご参加の場合
特 割: 弊社プロジェクト実施中、継続教育等の場合ご相談下さい
定 員: リモート定員 60名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
開催要件:
1.リモート参加要件
会社からの場合、個室(会議室等)から会社PCで御参加となります。
ご自宅からの場合、会社PCでのご参加となります。
その他チェックリストを事前にお渡しし環境をご確認頂きます。
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.FDAがCAPAに注目している理由
警告書の傾向分析
2.なぜ品質問題は再発するのか
直接原因と根本原因の相違
再発品質不良の根本原因
3.規制・規格を正しく理解する
CAPAの目的の理解
是正、予防、水平展開
( 是正した後、予防は有り得ない )
統計的手法を使用し最初に予防
予防するシステムを理解する
4.TQMの仕組みが不可欠
トップがマネジメントするシステム
PDCAをまわす
プロセス改善ツール
5.リスクベースプロセスアプローチ
9001,13485CAPA
リスクベースプロセスアプローチとは?
測定、分析、改善
6.用語を正しく理解する
修正、是正、予防の相違
逸脱、不適合、逸脱許可と特別採用の相違
7.修正、是正、予防プロセス
受入アクセプタンス活動
工程内アクセプタンス活動
最終出荷アクセプタンス活動
苦情処理プロセス
設計変更プロセス
逸脱不適合プロセス
NCフローチャート
修理プロセス
8.CAPAプロセスエスカレーション
CA・PAエスカレーションの仕組み
リスク判断基準
9.CAPA7ステップ
Step 1 問題を検出し明確にする
Step 2 問題の原因を詳しく調査する
Step 3 直接、根本原因の特定
Step 4 処置の特定と検証または妥当性確認
Step 5 予防または是正する為の処置
Step 6 処置の結果を文書化
Step 7 有効性の確認
10.フィードバック
フィードバックプロセス
CAPAはシステム改善プロセス
システムの有効性
11.品質問題の事例に学ぶ
Warning Letter Case Study
実際の事例

FDA MDR&CAR リモートコース Rev.11
リモート形式
〜 査察指摘リスクが高い行政報告プロセス 〜
21CFR : Part 820, 803, 806, ISO13485
このコースの売り上げ金の一部は人道支援活動に寄付されます
☆ 開催概要 ☆
新しいQSRに変わるQMSRでISO13485にないプロセスを学び早期にプロセスを構築する必要があります。FDA査察レポートの解析からワースト4位(全体の約10%)に急浮上しているMDR(有害事象報告)、日本でも Warning Letter (警告書)を受けているほとんどの企業がMDRの指摘を受けています。昨今のデータ偽装事件を受け、FDAはこれらにフォーカスし厳しく査察を行っています。これはMDR報告の判断に迷ったら報告するという米国の文化とは異なり、日本のメーカーは情報を積極的に発信しない隠すという古い会社の文化が根強くあることが要因の一つですが、そもそも規制を理解せず正しい判断を行っていないことが大きな原因です。医療機器を作っているメーカーは、患者さんの命を守るという社会的な責任があります。会社を守っているようでも、結果的に会社が倒産に追い込まれることもある非常にリスクの高いプロセスであることを理解すべきです。それには規制の正しい理解、規制を守る仕組みと組織が必要です。このセミナーでは、CAPAサブシステム(苦情、不適合、CAPA)の各プロセスとリンクし、日本のメーカーの Warning Letter (警告書)を引き合いに正しいMDRプロセスと関連するCAR(リコール)プロセスを構築することを理解します。
このコースはオンサイトでも実施しています。お問い合わせはこちらから。
日 時:2024年10月21日(月曜)9:00 〜 16:30
10月11日(金曜)締切
会 場:リモート(ZOOM)のみ
( ZOOMはブラズザからログインできます )
対 象:既に医療機器・IVDを米国に輸出している企業
これから医療機器・IVDを米国輸出する準備を行っている企業
既にFDA査察を終え対応に困っている企業
受講料: ¥55,000-( 消費税込み、テキスト代含む、ご来場の方には昼食代含まれます )
リモートの方には事前にテキストを郵送させて頂きます
割 引: ¥44,000-( 消費税込み、テキスト代含む、ご来場の方には昼食代含まれます )
1社2名以上ご出席の場合
定 員: リモート(30名)
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
特 割:弊社プロジェクトを実施中、又は継続研修をご希望の場合、御負担軽減のためさらに
割引をさせて頂きますのでお問い合わせください。この場合、正規料金でお申込の方
が優先され、出席できない場合がございますので予めご了承ください。
再受講:再受講をご希望の方はさらに割引が適用され、空席がある場合ご出席頂けます。
併せて過去ご出席なさった日をお知らせ下さい。
お申込:セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
開催要件:リモート参加の場合、リモート環境に確認とNDAへのサインをお願いしています。
折り返し、リモートリンクお振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.規制を理解する
過去事例と統計からみる規制違反
理解すべき規制・規格・ガイダンス
eMDR規制
苦情組織と力量
規制が求めるあるべき組織と力量
プロセス構成
2.有害事象報告( MDR )Part 803
MDR規制の基本要件
FDA Warning letter から
規制の目的、重要性
MDRとして報告すべき事象とは
2.1 定義を理解する
重篤な障害
機能不全
報告すべき Malfunction
合理的に推測する
認識する
2.2 4.5日、30日報告
報告形態と時間軸
5日報告
ディシジョンツリー
2.3 FDAへの報告
報告フロー
報告様式 3500A
報告不要の場合
報告書の種類
報告免除
Warning letter 警告書 !
2.4 MDRプロセス
MDR報告フロー
CA,PAプロセス
3.回収報告(CAR)Part 806
規制の理解
報告免除
定義
リコールフロー
任意と強制リコール
定期メンテナンスとリコール
3.1 リコール判断基準
リコール判断基準例
ラベリングの定義
ラベリング違反と法的根拠
報告すべきラベリング違反例
3.2 CAR ディシジョンツリー
リコールディシジョンツリー
3.3 報告書と処理フロー
リコール判断後フロー
リコールクラス
有効性の確認
CAPAとの関係
4.警告書から理解すべき注意点とプロセスへの展開
Warning Letter ( 警告書 )の事例から理解する
警告書から理解すべき注意点とプロセスへの展開
プロセス構築後のレトロスペクティブな是正とは
*コース内容が一部変更される場合がございますが大きな変更はございません。

FDA 設計管理2日間HBコース Rev.27
〜 安全性と性能を証明するフレームワーク 〜
NEW QMSR 対応 21CFR Part 820.30 / 75 ISO13485
QSR内部監査員コースご出席要件コースです
( オンサイト及び過去弊社コースを受講された方も要件を満たします )
会場参加、リモート、ハイブリッド形式で開催します
☆ 開催概要 ☆
ほとんどの日本企業に於ける医療機器開発・設計管理の仕組みは世界標準にほど遠く、これはグローバルに進出しようとする企業のみならず、患者の命を預かる全ての医療機器メーカーにとって大きなリスクを伴います。米国FDAは自らの査察で、医療機器品質不良の根本原因は上流側の設計管理にあると査察の結果から統計的に見抜いており、それはクオリス・イノーバの20年以上にわたるGAP監査でも明らかです。そこで本コースでは、グローバルな開発、設計管理を米国FDA「設計管理ガイダンス」及び欧米で取り入れられている開発フレームワークを元に体系的に解説、また、品質不良を発生させない仕組み(ロバスト設計)をタグチメソッドとライフサイクルマネジメントを用いて解説します。 2日目はプロセスバリデーションの本質、工程内試験に頼らない均一で高品質な製品を一貫して製造する為、パラメータによる製造方法を実現する工程設計について解説します。 ”規制” だからではなく、設計品質を高めることにより患者さんの命を守り、企業にとっても大きな潜在的利益を得られるという大原則は、エンジニアの方々から多くの共感と支持を頂いてきたクオリス・イノーバの最も人気のある価値あるコースです。 このコースはオンサイトでも実施しております。お問い合わせはこちらから。
日 時: 2024年10月24日(木曜)、25日(金曜) 9:30〜16:30
締 切: 2024年10月18日(金曜)
会 場: 全国町村会館 大ホール(東京・永田町) 永田町駅から徒歩1分( 3番出口 )
リモートで参加はZOOMを使用
講 師: クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象: 医療機器・IVD市場にこれから参入しようとしている企業
医療機器・IVDを海外に輸出しようとしている企業
FDA査察準備、MDSAPを受審しようと準備している企業
既にFDA査察を終え、指摘対応に苦慮している企業
製品実現に関わる各組織( マーケティング、開発、生産技術、品証など )
受講料: ¥99,000( 税込、テキスト・昼食含む )
割 引: ¥88,000( 税込、テキスト・昼食含む )
1社2名以上受講される場合
特 割: 再受講の方、プロジェクト終了企業様で継続研修としてご検討の方ご相談下さい。
定 員: クラス 定員 20名
リモート定員 30名
出席者全員に教育記録として利用できる修了書をお渡しします
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡いたします。
2名様以上ご参加の場合、お名前とローマ字読み、メールアドレスを連絡欄にご記入下さい。
持参品: 弊社QSR翻訳本第5版をご持参下さい。
お持ちでない方はこちらから特別価格でお求め頂けます。
ご注意: 本コースは、品質マネジメントシステムの一部を解説するため、システム全体の知識がないと理解できません。本コース受講前に9月開催QMSR2日間コースの受講をお勧めします。( QMSR2日間コースで訳本を配布しています )
開催要件:
リモートご参加の場合
ZOOM以外での対応はしておりません
会社からご参加の場合、会議室から会社PCでのご参加となります。
自宅からご参加の場合、会社PCでのご参加となり、
その他、チェックリストでご確認頂く為、事前にご確認をお願いしています。
メールでお問い合わせ
電話でお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1日目(セミナー内容)
A.コンプライアンスとインテグリティー
設計不良の原因は何か
設計CAPAとは
ケーススタディ
不正を排除する仕組み
規制・規格に満足するればOKか?
人の命
行政によるミッションの相違
B.医療機器規制を理解する
新しい規則QMSRと今後の対応値
適用規制、規格、ガイダンス
1.Subpart C 設計管理 820.30 ISO13485 - 7.3
設計管理の目的とその重要性
プロセスアプローチの枠組と製品実現フレームワークの重要性
ライフサイクルマネジメント(LCM)と Waterfall Model
関連する ISO13485 の項目(プロセス)
Form 483 の FY 統計的解析から理解すること
2.820.30 (a) 総則 ISO13485 - 7.3.1
プロセスアプローチ ISO13485 4.1 の理解と
経営者の責任(リソースの提供とKPI)
製品実現プロセス
タートルチャート
リソースとしての力量(教育と訓練) 820.25
文書体系から俯瞰する設計管理プロセス
3.820.30 (b) 設計及び開発計画 ISO13485 - 7.3.2
医療機器開発ライフサイクルマネジメント(LCM)
設計品質確立の重要な要素; コンカレントエンジニアリング(CE)
PM、PLの役割と責任
ライフサイクルリスクマネジメントの理解(LCRM) ISO14971
タグチメソッド開発フレームワークによるフロントローディング手法
マーケティング戦略の重要性
開発計画書
4.820.30 (c) 設計インプット ISO13485 - 7.3.3
コミュニケーションエラー
曖昧なニーズの明確化
要求仕様から完成品までの一貫性
トレーサビリティーマトリックス(TMX)
ラベリング、包装設計のインプット
820.120 機器のラベリング
820.130 機器の包装
5.820.30 (d) 設計アウトプット ISO13485 - 7.3.4
タートルチャートのアウトプット
設計アウトプットのリスト
E−BOMの役割
製造仕様書とは
6.820.30 (e) 設計レビュー ISO13485 - 7.3.5
問題の早期発見、解決が目的でセレモニーで終わらせない仕組み
設計品質確立の重要な要素
設計レビューとテクニカルレビュー
レビューワーと責任と権限
7.820.30 (f) 設計検証 ISO13485 - 7.3.6
インプットからアウトプットまでのトレーサビリティー
製品の安全性、性能を証明するエビデンスの作り方
設計検証計画に必要な要素
設計検証レポートはデータの信頼性を確保
8.820.30 (g) 設計バリデーション ISO13485 - 7.3.7
設計検証と設計バリデーションの相違
設計バリデーションの手法
2つのバリデーションの相違と記録
ソフトウェアのバリデーションとVモデル
3種類のバリデーションの相違
9.820.30 (h) 設計移管 ISO13485 - 7.3.8 → 第2日目(工程設計)
10.820.30 (i ) 設計変更 ISO13485 - 7.3.9
変更管理プロセスに伴う設計変更
ライフサイクルリスクマネジメント
タートルチャート
設計変更フロー
11.820.30 (j ) DHF ISO13485 - 7.3.10 設計・開発ファイル
DHF / DMR / DHR の定義
識別、ファイルの基本
文書体系と設計標準
2日目(セミナー内容)
設計移管と工程設計
820.30 (h)、13485 - 7.3.8、7.5.6
1.820.30 (h) 設計移管 ISO13485 - 7.3.8
ライフサイクルリスクマネジメントにおける設計移管(工程設計)
均一な製品を一貫して生産する手法
プロセスバリデーションの目的
2.なぜ工程バリデーションを実施するのか 820.75, ISO13485 - 7.5.6
プロセスバリデーションの目的とその本質
プロセスバリデーションの効果
工程管理、監視の目的と本質
医療機器規制・規格の理解
3.バリデーションすべき工程とは
バリデーションすべき工程の識別
検証工程とバリデーション工程の相違
設備バリデーションと工程バリデーション
バリデーション工程例
4.バリデーションの種類
バリデーションの種類
設備バリデーションと劣化パラメータ 820.70(g), 72(a)
コンピューターシステムバリデーション(CSV) 820.70(i), ISO13485 - 7.5.6 (g)
5.バリデーションステップ 820.75, ISO13485 - 7.5.6
VMP:バリデーションマスタープラン
DQ: 設計時適格性評価
IQ: 据付時適格性評価
OQ: 稼働時適格性評価
PQ: 製造時適格性評価
PPQ: プロセス街道性評価
バリデーション文書化
プロセスバリデーションの明確化
自動化工程の明確化
CSVカテゴリーの明確化
ソフトウェアのバリデーションの基本
リスクベースCSV
6.工程設計とプロセスバリデーション 820.30 (h)、 ISO13485 - 7.3.8
ライフサイクルマネジメントに於ける工程設計
工程設計の進め方
工程設計とプロセスリスク分析( PFMEA )
環境管理 820.70(c)、 ISO13485 - 6.4
汚染管理 820.70(e)、 ISO13485 - 6.4
副資材 820.70(h)、 ISO13485 - 7.5.2
工程設計PTMX
QC工程表
7.回顧的バリデーション
8.バリデーションのアウトプット
QC工程表と統計的手法(工程能力)
リバリデーション設定
設備インスペクションとメンテナンス
( 内容が変更される場合がございます )

FDA QMSR/QSR/13485
2日間ハイブリッドコース
〜 医療機器・IVDに求められるMSの本質を学ぶ 〜
21CFR Part 820 / ISO13485 / ( ISO9001 )
このコースは会場、リモートのハイブリッド開催です。
2024年2月に公示された QSR に変わる新たな規則 QMSR について、
ISO13485 との相違はもとより、原点に立ち返る MS の本質について学ぶ新たなコースです。
年末に実施する監査員コースの受講要件となります。
☆ 開催概要 ☆
QSR翻訳本(第5版)+QMSR翻訳本(第1版)付き
『 医療機器を作ることと、安全性と性能を証明することは異なる。』このことに経営陣が気付かない医療機器メーカーが規模の大小に関わらず法違反や患者の命に関わる事件を起こしています。 規制だけ満足していればよいという考え方が、どれだけこのビジネスでは会社のみならず社会にリスクがあるかのを最初に理解すべきです。
他方、市場を考えると、米国が最も大きなマーケットであることは医療機器でも例外では無く、米国での成功がグローバルを制すると言っても過言ではありません。しかし、米国で製品を上市するには医療機器規制 QMSR に基づく MS を構築し、『安全性と性能』を証明しなければなりません。2026年2月から新たに適用となるこの規則は、FDA が QSR を計画的に反映させた ISO13485(2016) が基本となります。今後は QSR の欠点でもあった MS の本質、リスクベースプロセスアプローチのフレームワークが必須となります。弊社の経験上 ISO1345 取得済みの企業であっても ISO に準拠しているとはとても言いがたく、FDA の査察アプローチは ISO と異なる為、 QMSR には準拠できません。今まで弊社は一貫して規制の本質を解き、事業活動とのインテグレーションを取ることにより、結果の出せるマネジメントシステムを提唱し、多くの企業の皆様の共感を集め支持されて参りました。米国に進出する企業のみならず、医療機器・IVDを開発しようとしている企業の皆様のマインドが大きく変わる価値あるクオリス・イノーバのメインコースです。
新規制が適用となるまで QSR が適用となる為、本コースでは現行 QSR, QSMR, ISO13485 について、リスクベースプロセスアプローチ、LCMといったシステムの根幹となるフレームワークを中心に概要を説明します。
オンサイト(一定人数が集まった会社でのセミナー)も可能です。お問合せはこちらから。
日 時: 2024年9月24日(火曜)、25日(水曜) 9:30〜16:30
締 切: 9月13日(金曜)
会 場: 全国町村会館(東京・永田町)(永田町駅から徒歩1分)
リモートで参加なさる場合、ZOOMでの御参加となります。
講 師: クオリス・イノーバ株式会社
代表取締役社長 木村 浩実
対 象: 医療機器・IVD市場にこれから参入しようと考えている企業
米国に輸出を計画している企業
米国FDA 査察準備を考えている企業
ODM、OEM として医療機器を受託しようとしている企業
受講料: ¥99,000−(税込)( 2日分、テキスト、対訳本2冊、昼食代含 )
リモートで参加なさる方には事前にテキスト、対訳本を郵送いたします。
昼食は会場ご出席者のみ
割 引: ¥88,000−(税込)
1社2名以上ご参加の場合
特 割: 企業で行うQMS構築プロジェクトなどや企業で弊社が実施した継続研修などで
ご利用の場合、特別割引を適用させて頂きますのでメールでご相談下さい。
満席となった場合、正規料金をお支払いの方が優先となりご希望に添えない場合
がございますので予めご了承ください。
定 員: クラス 定員 20名様
リモート定員 30名様
( 出席者全員に教育記録として利用できる修了書をお渡しします )
開催要件:
1.リモート参加要件
会社からの場合、個室(会議室等)から会社PCで御参加となります。
ご自宅からの場合、会社PCでのご参加となります。
その他チェックリストを事前にお渡しし環境をご確認頂きます。
2.COVID19対策
弊社のウィルス対策を順守して頂きます。
当日発熱がある場合、参加を御控え頂きます。
お申込: セミナー申込みページからお申し込み下さい。
折り返し、会場、お振り込み先情報などをメールでご案内致します。
2名以上で参加なさる方ははその旨ご記入下さい。
割引料金でご案内致します。
![]() メールでのお問い合わせ
メールでのお問い合わせ
![]() 電話でのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.医療機器を作ることと、安全性、性能を証明することは異なる
なぜ承認が取得できないのか
なぜ査察で警告を受け裁判にまで発展するのか
2.コンプライアンスとインテグリティー
患者の命と会社の危機管理
データインテグリティー
3.医療機器規制の理解
行政によるミッションの相違
米国規制法
医療機器規制の理解
QSR vs QMSR
4.FDA査察
FDA査察の最新動向とISO企業の実態
FDA査察、頻度、査察タイプ
QSITアプローチ
A リスクベースプロセスアプローチ
Subpart_B 品質システム要求事項
820.20 経営者の責任
プロセスアプローチ
タートルチャート
マネジメントレビュー
文書体系
品質監査
力量
Subpart_D 文書管理
820.40 文書管理
Subpart_O 統計的手法
820.250 統計的手法
Subpart_J 是正及び予防処置
820.100 CAPA
用語の正しい理解
エスカレーション
プロセスアプローチ
B 製品実現フレームワーク
Subpart_C 設計管理
Subpart_E 購買管理
Subpart_F 識別及びトレーサビリティー
Subpart_G 生産及び工程管理
Subpart_H アクセプランスアクティビティー
Subpart_I 不適合製品
Subpart_K ラベリング及び包装管理
Subpart_L 取扱い、保管、流通及び据付け
Subpart_M 記録
Subpart_N 附帯サービス
リスクベースプロセスアプローチ
〜 QMSの原則を学ぶ 〜
FDA QMSR / ISO13485 / ISO9001
このコースの売上金の一部は人道支援活動に寄付されます
☆ 開催概要 ☆
実は経営の本質がISO QMSであることを経営者は誰も気付かない、ISOの監査員は誰も指摘しない。事件を起こし取り返しのつかない事態になって初めてQMSが原則であることを学ぶ。プロセスアプローチの仕組みを知らないため、組織論がどうだとか嘯き、組織はもの造りのフレームを成していない。さらに、プロセスの説明責任を持つプロセスオーナーにその自覚が無く、規制、規格など適当にやっていればいいと指示した結果、FDAから深刻な警告書を受け裁判沙汰となる。QMSの原則を理解しているかどうかは簡単な質問でわかってしまう。FDA QSRとQMSRの相違はまさにこのパート。QMSRベースの査察では、このパートがキーとなります。エキスパートが見抜くシステムの悪さ加減は、プロセスアプローチの理解にあります。
日 時: 2024年9月2日(月曜)開催 13:00〜17:00
締 切: 2024年8月26日(月曜)
会 場: リモート (ZOOMのみ)
講 師: クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
受講料: ¥33,000( 税込み、テキスト )
割引き: ¥27,500( 税込み、テキスト )1社2名以上ご参加
定 員: 50名
出席者全員に修了書を発行します
お申込: セミナーお申込ページよりお申し込み下さい
![]()
メールでお問い合わせ
電話でお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.医療機器を作ることと、安全性、性能を証明することは異なる
− なぜ承認が取得できなかったのか
− なぜ査察で警告を受けてしまったのか
2.安全性、性能の説明責任を果たすフレームワーク
− QMS規則は最小要件
− プロセスアプローチを見れば企業の姿勢がよくわかる
3.QMSの7原則
− ビジネスマネジメントの原則
− ビジネスの結果を出すツール
− 組織では無くプロセスファースト
4.プロセスとは何か
− プロセスの定義
− システムを構成するプロセス
− 製品実現の為のプロセス相互関係
− タートルチャート
− プロセスの運用管理
5.リスクベースプロセスアプローチ
− ISO13485 4.1.2 の理解図
− プロセスの監視測定 8.2.5
− 製品実現フレームワークの為のプロセス
6.システムパフォーマンス
− 適切性、妥当性、有効性のトップマネジメントへの報告
− プロセスの文書化
− 内部コミュニケーション
− プロセスの監視測定
7.フィードバックプロセスと、分析
− フィードバックデータとは
− QSR,ガイダンスで決められたデータソース
− データを分析して予防する
− プロセスの改善はプロセスオーナーの責任
8.プロセスを改善する為のCAPA
− 問題を起こさないための予防活動
− CAPAはプロセスにコミットするオーナーの判断
9.内部監査はプロセスアプローチ
− 監査はプロセス毎に実施
− トップマネジメントへの報告

FDA 設計管理2日間HBコース Rev.27
〜 安全性と性能の説明責任を果たすフレームワーク 〜
NEW QMSR 対応 21CFR Part 820.30 / 75 ISO13485
QSR内部監査員コースご出席要件コースです
( オンサイト及び過去弊社コースを受講された方も要件を満たします )
会場参加、リモート、ハイブリッド形式で開催します
☆ 開催概要 ☆
ほとんどの日本企業に於ける医療機器開発・設計管理の仕組みは世界標準にほど遠く、これはグローバルに進出しようとする企業のみならず、患者の命を預かる全ての医療機器メーカーにとって大きなリスクを伴います。米国FDAは自らの査察で、医療機器品質不良の根本原因は上流側の設計管理にあると査察の結果から統計的に見抜いており、それはクオリス・イノーバの20年以上にわたるGAP監査でも明らかです。そこで本コースでは、グローバルな開発、設計管理を米国FDA「設計管理ガイダンス」及び欧米で取り入れられている開発フレームワークを元に体系的に解説、また、品質不良を発生させない仕組み(ロバスト設計)をタグチメソッドとライフサイクルマネジメントを用いて解説します。 2日目はプロセスバリデーションの本質、工程内試験に頼らない均一で高品質な製品を一貫して製造する為、パラメータによる製造方法を実現する工程設計について解説します。 ”規制” だからではなく、設計品質を高めることにより患者さんの命を守り、企業にとっても大きな潜在的利益を得られるという大原則は、エンジニアの方々から多くの共感と支持を頂いてきたクオリス・イノーバの最も人気のある価値あるコースです。 このコースはオンサイトでも実施しております。お問い合わせはこちらから。
日 時: 2024年7月1日(月曜)、2日(火曜) 9:30〜16:30
締 切: 2024年6月28日(金曜)
会 場: 全国町村会館 大ホール(東京・永田町) 永田町駅から徒歩1分( 3番出口 )
リモートで参加はZOOMを使用
講 師: クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象: 医療機器・IVD市場にこれから参入しようとしている企業
医療機器・IVDを海外に輸出しようとしている企業
FDA査察準備、MDSAPを受審しようと準備している企業
既にFDA査察を終え、指摘対応に苦慮している企業
製品実現に関わる各組織( マーケティング、開発、生産技術、品証など )
受講料: ¥99,000( 税込、テキスト・昼食含む )
割 引: ¥88,000( 税込、テキスト・昼食含む )
1社2名以上受講される場合
特 割: 再受講の方、プロジェクト終了企業様で継続研修としてご検討の方ご相談下さい。
定 員: クラス 定員 20名
リモート定員 30名
出席者全員に教育記録として利用できる修了書をお渡しします
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡いたします。
2名様以上ご参加の場合、お名前とローマ字読み、メールアドレスを連絡欄にご記入下さい。
持参品: 弊社QSR翻訳本第5版をご持参下さい。
お持ちでない方はこちらから特別価格でお求め頂けます。
ご注意: 本コースは、品質マネジメントシステムの一部を解説するため、システム全体の知識がないと理解できません。本コース受講前に10月開催QMSR2日間コースの受講をお勧めします。( QMSR2日間コースで訳本を配布しています )
開催要件:
1.コロナウイルス対応
弊社のウィルス対策を順守して戴きます。
当日熱がある場合、会場への出席はお控え頂きます。
2.リモートご参加要件
ZOOM以外での対応はしておりません
会社からご参加の場合、会議室から会社PCでのご参加となります。
自宅からご参加の場合、会社PCでのご参加となります。
その他、チェックリストでご確認頂く為、事前にご確認をお願いしています。
メールでお問い合わせ
電話でお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1日目(セミナー内容)
A.コンプライアンスとインテグリティー
設計不良の原因は何か
設計CAPAとは
ケーススタディ
不正を排除する仕組み
規制・規格に満足するればOKか?
人の命
行政によるミッションの相違
B.医療機器規制を理解する
新しい規則QMSRと今後の対応値
適用規制、規格、ガイダンス
1.Subpart C 設計管理 820.30 ISO13485 - 7.3
設計管理の目的とその重要性
プロセスアプローチの枠組と製品実現フレームワークの重要性
ライフサイクルマネジメント(LCM)と Waterfall Model
関連する ISO13485 の項目(プロセス)
Form 483 の FY 統計的解析から理解すること
2.820.30 (a) 総則 ISO13485 - 7.3.1
プロセスアプローチ ISO13485 4.1 の理解と
経営者の責任(リソースの提供とKPI)
製品実現プロセス
タートルチャート
リソースとしての力量(教育と訓練) 820.25
文書体系から俯瞰する設計管理プロセス
3.820.30 (b) 設計及び開発計画 ISO13485 - 7.3.2
医療機器開発ライフサイクルマネジメント(LCM)
設計品質確立の重要な要素; コンカレントエンジニアリング(CE)
PM、PLの役割と責任
ライフサイクルリスクマネジメントの理解(LCRM) ISO14971
タグチメソッド開発フレームワークによるフロントローディング手法
マーケティング戦略の重要性
開発計画書
4.820.30 (c) 設計インプット ISO13485 - 7.3.3
コミュニケーションエラー
曖昧なニーズの明確化
要求仕様から完成品までの一貫性
トレーサビリティーマトリックス(TMX)
ラベリング、包装設計のインプット
820.120 機器のラベリング
820.130 機器の包装
5.820.30 (d) 設計アウトプット ISO13485 - 7.3.4
タートルチャートのアウトプット
設計アウトプットのリスト
E−BOMの役割
製造仕様書とは
6.820.30 (e) 設計レビュー ISO13485 - 7.3.5
問題の早期発見、解決が目的でセレモニーで終わらせない仕組み
設計品質確立の重要な要素
設計レビューとテクニカルレビュー
レビューワーと責任と権限
7.820.30 (f) 設計検証 ISO13485 - 7.3.6
インプットからアウトプットまでのトレーサビリティー
製品の安全性、性能を証明するエビデンスの作り方
設計検証計画に必要な要素
設計検証レポートはデータの信頼性を確保
8.820.30 (g) 設計バリデーション ISO13485 - 7.3.7
設計検証と設計バリデーションの相違
設計バリデーションの手法
2つのバリデーションの相違と記録
ソフトウェアのバリデーションとVモデル
3種類のバリデーションの相違
9.820.30 (h) 設計移管 ISO13485 - 7.3.8 → 第2日目(工程設計)
10.820.30 (i ) 設計変更 ISO13485 - 7.3.9
変更管理プロセスに伴う設計変更
ライフサイクルリスクマネジメント
タートルチャート
設計変更フロー
11.820.30 (j ) DHF ISO13485 - 7.3.10 設計・開発ファイル
DHF / DMR / DHR の定義
識別、ファイルの基本
文書体系と設計標準
2日目(セミナー内容)
設計移管と工程設計
820.30 (h)、13485 - 7.3.8、7.5.6
1.820.30 (h) 設計移管 ISO13485 - 7.3.8
ライフサイクルリスクマネジメントにおける設計移管(工程設計)
均一な製品を一貫して生産する手法
プロセスバリデーションの目的
2.なぜ工程バリデーションを実施するのか 820.75, ISO13485 - 7.5.6
プロセスバリデーションの目的とその本質
プロセスバリデーションの効果
工程管理、監視の目的と本質
医療機器規制・規格の理解
3.バリデーションすべき工程とは
バリデーションすべき工程の識別
検証工程とバリデーション工程の相違
設備バリデーションと工程バリデーション
バリデーション工程例
4.バリデーションの種類
バリデーションの種類
設備バリデーションと劣化パラメータ 820.70(g), 72(a)
コンピューターシステムバリデーション(CSV) 820.70(i), ISO13485 - 7.5.6 (g)
5.バリデーションステップ 820.75, ISO13485 - 7.5.6
VMP:バリデーションマスタープラン
DQ: 設計時適格性評価
IQ: 据付時適格性評価
OQ: 稼働時適格性評価
PQ: 製造時適格性評価
PPQ: プロセス街道性評価
バリデーション文書化
プロセスバリデーションの明確化
自動化工程の明確化
CSVカテゴリーの明確化
ソフトウェアのバリデーションの基本
リスクベースCSV
6.工程設計とプロセスバリデーション 820.30 (h)、 ISO13485 - 7.3.8
ライフサイクルマネジメントに於ける工程設計
工程設計の進め方
工程設計とプロセスリスク分析( PFMEA )
環境管理 820.70(c)、 ISO13485 - 6.4
汚染管理 820.70(e)、 ISO13485 - 6.4
副資材 820.70(h)、 ISO13485 - 7.5.2
工程設計PTMX
QC工程表
7.回顧的バリデーション
8.バリデーションのアウトプット
QC工程表と統計的手法(工程能力)
リバリデーション設定
設備インスペクションとメンテナンス
( 内容が変更される場合がございます )

〜 FDA査察指摘ワースト&キープロセスを理解する 〜
NEW QMSR 対応 21CFR Part 820.100、ISO13485 8.5、ISO9001 10
このコースの売上金の一部は人道支援に寄付されます
* コース概要 *
CAPAプロセスを確認するだけで、その会社の品質システムや品質に対する理解度がよくわかります。CAPAプロセスに問題があれば、品質マネジメントシステムの適切性、妥当性はなく、特にマネジメントレビューがまともに運用されているとは思われません。品質マネジメントシステムの原則はプロセスアプローチ、PDCAをまわす為、マネジメントレビューのインプットでプロセスを監視し、改善ツールであるCAPAでプロセスを改善します。当たり前のことがシステムとして運用されていないことを理解している規制当局は、CAPAプロセスにフォーカスして確認しようとします。QMSが機能していないことは昨今の品質に関わる事件から明らかで、医療機器・IVDの場合、命に関わる大問題です。本コースでは今までの内容を大きく変更し、プロセスアプローチの手法に重点を置き、事例を元にCAPAの本質を理解して頂きます。 このコースは受講者と同じ目線の御社事例を用いることでより理解が高まるオンサイトセミナーが可能です。お問合せはこちらから。
日 時:2024年6月24日(月曜)9:30 〜 16:30
会 場:リモートのみ
( 参加はZOOMのみとなります )
講 師:クオリス・イノーバ株式会社代表取締役社長 木村 浩実
対 象:医療機器、IVD、医薬品等に限定せず、CAPAプロセスを学びたい企業様
受講料: ¥55.000-( 税込、テキスト代 )
事前にテキストを郵送致します
割 引: ¥44.000-( 税込、テキスト代 )
1社2名様以上ご参加の場合
特 割: 弊社プロジェクト実施中、継続教育等の場合ご相談下さい
定 員: リモート定員 60名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
開催要件:
1.リモート参加要件
会社からの場合、個室(会議室等)から会社PCで御参加となります。
ご自宅からの場合、会社PCでのご参加となります。
その他チェックリストを事前にお渡しし環境をご確認頂きます。
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.FDAがCAPAに注目している理由
警告書の分析
2.なぜ品質問題は再発するのか
直接原因と根本原因の相違
再発品質不良の根本原因
3.規制・規格を正しく理解する
CAPAの目的の理解
是正、予防、水平展開
統計的手法を使用し最初に予防
予防するシステムを理解する
4.TQMの仕組みが不可欠
トップがマネジメントするシステム
PDCAをまわす
プロセス改善ツール
5.リスクベースプロセスアプローチ
9001,13485CAPA
リスクベースプロセスアプローチとは?
測定、分析、改善
6.用語を正しく理解する
修正、是正、予防の相違
逸脱、不適合、逸脱許可と特別採用の相違
7.修正、是正、予防プロセス
受入アクセプタンス活動
工程内アクセプタンス活動
最終出荷アクセプタンス活動
苦情処理プロセス
設計変更プロセス
逸脱不適合プロセス
NCフローチャート
修理プロセス
8.CAPAプロセスエスカレーション
CA・PAエスカレーションの仕組み
リスク判断基準
9.CAPA7ステップ
Step 1 問題を検出し明確にする
Step 2 問題の原因を詳しく調査する
Step 3 根本原因の特定
Step 4 処置の特定と検証または妥当性確認
Step 5 予防または是正する為の処置
Step 6 処置の結果を文書化
Step 7 有効性の確認
10.フィードバック
フィードバックプロセス
CAPAはシステム改善プロセス
システムの有効性
11.品質問題の事例に学ぶ
Warning Letter Case Study
実際の事例

購買プロセス リモートコース
〜 FDA 査察指摘 Worst 5 購買プロセスの本質と対策 〜
21CFR Part 820・ISO13485・ISO9001・ガイダンス
このコースの売り上げ金の一部は人道支援活動に寄付されます
リモートで開催され会場での開催はありません
☆ 開催概要 ☆
FDA査察で必ず指摘を受ける(ワースト5)重要なプロセス。アッセンブラーが多くなった昨今、行政当局も購買プロセスに多くの査察時間を費やすようになりました。また、重要構成品のアウトソースを行う企業がほとんどで、実際はアウトソースとして扱うべきところを、パーツサプライヤとして扱ってみたり、マネジメントシステムの傘下として扱うべきサービス購買の要求事項を理解していません。さらに、購買プロセスは、言われた通りにモノを買っていればよいというものではなく、サプライチェーン全体を俯瞰し、製品企画時の上流から積極的にコンカレントで参画、最も効率よいQCDを提案することこそ会社に貢献するプロセスとして価値があります。その為には、戦略的な組織体制とフレームを構築する必要があり、開発購買や情報購買と言ったフレームワークは早期の製品ローンチを実現します。新たな規制QMSRを想定し、ISO13485、9001の要求も加味しながら理解を深めます。
オンサイト(御社のみで)でも可能です。お問い合わせはこちらから。
日 時:2024年6月10日(月曜) 9:30 〜 16:30
会 場:リモートのみ
( リモートは ZOOM でのご参加となります )
講 師:クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
ODM、OEMとして設計・製造を受託している企業
受講料: ¥55.000-( 消費税込、テキスト事前郵送 )
割 引: ¥44.000-( 消費税込、テキスト事前郵送 )
1社2名様以上ご参加の場合
特 割: 弊社プロジェクト実施中、継続教育等の場合ご相談下さい。
定 員: リモート 定員 40名
( 出席者全員に教育記録として利用できる修了書をお渡しします )
申込締切: 5月31日(金曜)
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
![]()
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
A.FDA規制、査察の最新情報と傾向分析(QMSRの動向)
1.適用規制・規格
購買プロセスへの規制
ガイダンスから学ぶ
2.プロセスアプローチの理解
重要構成プロセスであることの理解
プロセスの役割
3.購買管理の目的
購買プロセスの目的
製品購買とサービス購買
購買製品の検証(受入検査)
4.用語の定義
製品購買、サービス購買とは
プロセスのアウトソースと部品購買の相違
アウトソース先の管理、監視
5.購買管理
GHTF( IMDRF )ガイダンスに基づく手順
購買管理の7ステップ
6.購買方針
購買方針のない購買は不正を生む
7.戦略・開発購買
購買プロセスの製品企画への関わり方
戦略購買
開発購買
情報購買
開発スピードを上げ原価削減、購買活動のスピードを上げある
ベストプラクティスの収集と技術の取り込み
8.購買計画
製品企画段階から参画
供給する製品、サービスの特定
技術及びプロセス情報の特定
供給者候補の特定
リスク、管理方法の特定
9.供給者候補選定
供給者ビジネス、運用能力評価
供給者候補選定
10.供給者選定
評価選定基準の策定
供給業者候補とのコミュニケーション
供給者候補の能力評価
供給者の承認
11.管理方法の取り決め
購買情報とは
管理方法
購買製品の検証
12.変更時対応
変更管理規定
契約変更通知
4Mと変化点管理
13.納期、測定、監視
購買製品の監視QCD
リスクベースに応じた管理、監視
SQCを用いた分析、改善
金型管理
14.フィードバック、コミュニケーション
CR
SCAR
供給者評価シート
15.サプライヤー監査
リスクベース監査
5つの監査、評価エリア
情報購買、戦略上重要な監査チーム
16.物流(ロジスティクス)
取扱、倉庫、輸送管理
( 内容が変更になる場合もございますが大きな変更はございません。)

FDA QMSR/QSR/13485
2日間ハイブリッドコース
〜 医療機器・IVDに求められるMSの本質を学ぶ 〜
21CFR Part 820 / ISO13485 / ( ISO9001 )
このコースは会場、リモートのハイブリッド開催です。
2024年2月に公示された QSR に変わる新たな規則 QMSR について、
ISO13485 との相違はもとより、原点に立ち返る MS の本質について学ぶ新たなコースです。
年末に実施する監査員コースの受講要件となります。
☆ 開催概要 ☆
QSR翻訳本(第5版)+QMSR翻訳本(第1版)付き
『 医療機器を作ることと、安全性と性能を証明することは異なる。』このことに経営陣が気付かない医療機器メーカーが、規模の大小に関わらず、法違反や患者の命に関わる事件を起こしています。 規制だけ満足していればよいという考え方が、どれだけこのビジネスでは会社のみならず社会にリスクがあるかのを最初に理解すべきです。
他方、市場を考えると、米国が最も大きなマーケットであることは医療機器でも例外では無く、米国での成功がグローバルを制すると言っても過言ではありません。しかし、米国で製品を上市するには医療機器規制 QMSR に基づく MS を構築し、『安全性と性能』を証明しなければなりません。2026年2月から新たに適用となるこの規則は、FDA が QSR を計画的に反映させた ISO13485(2016) がほぼ準拠となります。今後は QSR の欠点でもあった MS の本質、リスクベースプロセスアプローチのフレームワークが必須となります。弊社の経験上 ISO1345 取得済みの企業であっても ISO に準拠しているとはとても言いがたく、FDA の査察アプローチは ISO と異なる為、 QMSR には準拠できませんのでご注意下さい。今まで弊社は一貫して規制の本質を解き、事業活動とのインテグレーションを取ることにより、結果の出せるマネジメントシステムを提唱し、多くの企業の皆様の共感を集め支持されて参りました。米国に進出する企業のみならず、医療機器・IVDを開発しようとしている企業の皆様のマインドが大きく変わる価値あるクオリス・イノーバのメインコースです。
新規制が適用となるまで QSR が適用となる為、本コースでは現行 QSR, QSMR, ISO13485 について、リスクベースプロセスアプローチ、製品実現フレームワークといったシステムの根幹となるフレームワークを中心に概要を説明します。
オンサイト(一定人数が集まった会社でのセミナー)も可能です。お問合せはこちらから。
日 時: 2024年5月27日(月曜)、28日(火曜) 9:30〜16:30
締 切: 5月17日(金曜)
会 場: 全国町村会館(東京・永田町)(永田町駅から徒歩1分)
リモートで参加なさる場合、ZOOMでの御参加となります。
講 師: クオリス・イノーバ株式会社
代表取締役社長 木村 浩実
対 象: 医療機器・IVD市場にこれから参入しようと考えている企業
米国に輸出を計画している企業
米国FDA 査察準備を考えている企業
ODM、OEM として医療機器を受託しようとしている企業
受講料: ¥99,000−(税込)( 2日分、テキスト、対訳本2冊、昼食代含 )
リモートで参加なさる方には事前にテキスト、対訳本を郵送いたします。
昼食は会場ご出席者のみ
割 引: ¥88,000−(税込)
1社2名以上ご参加の場合
特 割: 企業で行うQMS構築プロジェクトなどや企業で弊社が実施した継続研修などで
ご利用の場合、特別割引を適用させて頂きますのでメールでご相談下さい。
満席となった場合、正規料金をお支払いの方が優先となりご希望に添えない場合
がございますので予めご了承ください。
定 員: クラス 定員 20名様
リモート定員 30名様
( 出席者全員に教育記録として利用できる修了書をお渡しします )
開催要件:
1.リモート参加要件
会社からの場合、個室(会議室等)から会社PCで御参加となります。
ご自宅からの場合、会社PCでのご参加となります。
その他チェックリストを事前にお渡しし環境をご確認頂きます。
2.COVID19対策
弊社のウィルス対策を順守して頂きます。
当日発熱がある場合、参加を御控え頂きます。
お申込: セミナー申込みページからお申し込み下さい。
折り返し、会場、お振り込み先情報などをメールでご案内致します。
2名以上で参加なさる方ははその旨ご記入下さい。
割引料金でご案内致します。
![]() メールでのお問い合わせ
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.コンプライアンスとインテグリティー
患者の命と会社の危機管理
データインテグリティー
2.医療機器規制の理解
行政によるミッションの相違
米国規制法
医療機器規制の理解
QSR vs QMSR
3.FDA査察
ISO企業の実態
FDA査察、頻度、査察タイプ
QSITアプローチ
A リスクベースプロセスアプローチ
Subpart_B 品質システム要求事項
820.20 経営者の責任
プロセスアプローチ
タートルチャート
マネジメントレビュー
文書体系
品質監査
力量
820.20 経営者の責任
Subpart_D 文書管理
820.40 文書管理
Subpart_O 統計的手法
820.250 統計的手法
Subpart_J 是正及び予防処置
820.100 CAPA
用語の正しい理解
エスカレーション
プロセスアプローチ
B 製品実現フレームワーク
Subpart_C 設計管理
Subpart_E 購買管理
Subpart_F 識別及びトレーサビリティー
Subpart_G 生産及び工程管理
Subpart_H アクセプランスアクティビティー
Subpart_I 不適合製品
Subpart_K ラベリング及び包装管理
Subpart_L 取扱い、保管、流通及び据付け
Subpart_M 記録
Subpart_N 附帯サービス
FDA MDR&CAR リモートコース Rev.10
リモート形式
〜 査察指摘リスクが高い行政報告プロセス 〜
21CFR : Part 820, 803, 806, ISO13485
このコースの売り上げ金の一部は人道支援活動に寄付されます
☆ 開催概要 ☆
新しいQSRに変わるQMSRでISO13485にないプロセスを学び早期にプロセスを構築する必要があります。FDA査察レポートの解析からワースト4位(全体の約10%)に急浮上しているMDR(有害事象報告)、日本でも Warning Letter (警告書)を受けているほとんどの企業がMDRの指摘を受けています。昨今のデータ偽装事件を受け、FDAはこれらにフォーカスし厳しく査察を行っています。これはMDR報告の判断に迷ったら報告するという米国の文化とは異なり、日本のメーカーは情報を積極的に発信しない隠すという古い会社の文化が根強くあることが要因の一つですが、そもそも規制を理解せず正しい判断を行っていないことが大きな原因です。医療機器を作っているメーカーは、患者さんの命を守るという社会的な責任があります。会社を守っているようでも、結果的に会社が倒産に追い込まれることもある非常にリスクの高いプロセスであることを理解すべきです。それには規制の正しい理解、規制を守る仕組みと組織が必要です。このセミナーでは、CAPAサブシステム(苦情、不適合、CAPA)の各プロセスとリンクし、日本のメーカーの Warning Letter (警告書)を引き合いに正しいMDRプロセスと関連するCAR(リコール)プロセスを構築することを理解します。
このコースはオンサイトでも実施しています。お問い合わせはこちらから。
日 時:2024年3月4日(月曜)9:30 〜 16:30
会 場:リモート(ZOOM)のみ
( ZOOMはブラズザからログインできます )
対 象:既に医療機器・IVDを米国に輸出している企業
これから医療機器・IVDを米国輸出する準備を行っている企業
既にFDA査察を終え対応に困っている企業
受講料: ¥55,000-( 消費税込み、テキスト代含む、ご来場の方には昼食代含まれます )
リモートの方には事前にテキストを郵送させて頂きます
割 引: ¥44,000-( 消費税込み、テキスト代含む、ご来場の方には昼食代含まれます )
1社2名以上ご出席の場合
定 員: リモート(30名)
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
特 割:弊社プロジェクトを実施中、又は継続研修をご希望の場合、御負担軽減のためさらに
割引をさせて頂きますのでお問い合わせください。この場合、正規料金でお申込の方
が優先され、出席できない場合がございますので予めご了承ください。
再受講:再受講をご希望の方はさらに割引が適用され、空席がある場合ご出席頂けます。
併せて過去ご出席なさった日をお知らせ下さい。
お申込:セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
開催要件:リモート参加の場合、リモート環境に確認とNDAへのサインをお願いしています。
折り返し、リモートリンクお振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.規制を理解する
過去事例と統計からみる規制違反
理解すべき規制・規格・ガイダンス
eMDR規制
苦情組織と力量
規制が求めるあるべき組織と力量
プロセス構成
2.有害事象報告( MDR )Part 803
MDR規制の基本要件
FDA Warning letter から
規制の目的、重要性
MDRとして報告すべき事象とは
2.1 定義を理解する
重篤な障害
機能不全
報告すべき Malfunction
合理的に推測する
認識する
2.2 4.5日、30日報告
報告形態と時間軸
5日報告
ディシジョンツリー
2.3 FDAへの報告
報告フロー
報告様式 3500A
報告不要の場合
報告書の種類
報告免除
Warning letter 警告書
2.4 MDRプロセス
MDR報告フロー
CA,PAプロセス
3.回収報告(CAR)Part 806
規制の理解
報告免除
定義
リコールフロー
任意と強制リコール
定期メンテナンスとリコール
3.1 リコール判断基準
リコール判断基準例
ラベリングの定義
ラベリング違反と法的根拠
報告すべきラベリング違反例
3.2 CAR ディシジョンツリー
リコールディシジョンツリー
3.3 報告書と処理フロー
リコール判断後フロー
リコールクラス
有効性の確認
CAPAとの関係
4.警告書から理解すべき注意点とプロセスへの展開
Warning Letter ( 警告書 )の事例から理解する
警告書から理解すべき注意点とプロセスへの展開
プロセス構築後のレトロスペクティブな是正とは
*コース内容が一部変更される場合がございますが大きな変更はございません。

〜 FDA査察指摘ワースト&キープロセスを理解する 〜
NEW QMSR 対応 21CFR Part 820.100、ISO13485 8.5、ISO9001 10
このコースの売上金の一部は人道支援に寄付されます
* コース概要 *
CAPAプロセスを確認するだけで、その会社の品質システムや品質に対する理解度がよくわかります。CAPAプロセスに問題があれば、品質マネジメントシステムの適切性、妥当性はなく、特にマネジメントレビューがまともに運用されているとは到底思われません。品質マネジメントシステムの原則はプロセスアプローチ、PDCAをまわす為、マネジメントレビューのインプットでプロセスを監視し、改善ツールであるCAPAでプロセスを改善します。当たり前のことがシステムとして運用されていないことを理解している規制当局は、CAPAプロセスにフォーカスして確認しようとします。QMSが機能していないことは昨今の品質に関わる事件から明らかで、医療機器・IVDの場合、命に関わる大問題です。本コースでは今までの内容を大きく変更し、プロセスアプローチの手法に重点を置き、事例を元にCAPAの本質を理解して頂きます。 このコースは受講者と同じ目線の御社事例を用いることでより理解が高まるオンサイトセミナーが可能です。お問合せはこちらから。
日 時:2024年1月29日(月曜)9:30 〜 16:30
会 場:全国町村会館 大ホール(東京・永田町) 永田町駅から徒歩1分( 3番出口 )
リモートで参加はZOOMとなります
講 師:クオリス・イノーバ株式会社代表取締役社長 木村 浩実
対 象:医療機器、IVD、医薬品等に限定せず、CAPAプロセスを学びたい企業様
受講料: ¥55.000-( 税込、テキスト代 )
事前にテキストを郵送致します
割 引: ¥44.000-( 税込、テキスト代 )
1社2名様以上ご参加の場合
特 割: 弊社プロジェクト実施中、継続教育等の場合ご相談下さい
定 員: クラス 定員 20名
リモート定員 30名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
開催要件:
1.リモート参加要件
会社からの場合、個室(会議室等)から会社PCで御参加となります。
ご自宅からの場合、会社PCでのご参加となります。
その他チェックリストを事前にお渡しし環境をご確認頂きます。
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.FDAがCAPAに注目している理由
警告書の分析
2.なぜ品質問題は再発するのか
直接原因と根本原因の相違
再発品質不良の根本原因
3.規制・規格を正しく理解する
CAPAの目的の理解
是正、予防、水平展開
統計的手法を使用し最初に予防
予防するシステムを理解する
4.TQMの仕組みが不可欠
トップがマネジメントするシステム
PDCAをまわす
プロセス改善ツール
5.リスクベースプロセスアプローチ
9001,13485CAPA
リスクベースプロセスアプローチとは?
測定、分析、改善
6.用語を正しく理解する
修正、是正、予防の相違
逸脱、不適合、逸脱許可と特別採用の相違
7.修正、是正、予防プロセス
受入アクセプタンス活動
工程内アクセプタンス活動
最終出荷アクセプタンス活動
苦情処理プロセス
設計変更プロセス
逸脱不適合プロセス
NCフローチャート
修理プロセス
8.CAPAプロセスエスカレーション
CA・PAエスカレーションの仕組み
リスク判断基準
9.CAPA7ステップ
Step 1 問題を検出し明確にする
Step 2 問題の原因を詳しく調査する
Step 3 根本原因の特定
Step 4 処置の特定と検証または妥当性確認
Step 5 予防または是正する為の処置
Step 6 処置の結果を文書化
Step 7 有効性の確認
10.フィードバック
フィードバックプロセス
CAPAはシステム改善プロセス
システムの有効性
11.品質問題の事例に学ぶ
Warning Letter Case Study
実際の事例

FDA QSR/QMSR 監査員コース Rev.17
〜 QSR 対応内部監査員養成のための社内力量認定2日間教育コース 〜
* FDA QSIT をベースにプロセスアプローチで実施することを理解します *
このコースの売り上げ金の一部は人道支援活動に寄付されます
☆ 開催概要 ☆
FDAの監査資料の結果から、品質監査( Part 820.22 )の指摘は、もうすぐワースト10に入るほど急上昇しているセクションです。FDAは、いい加減な監査報告が経営者に報告されていることが根本原因であることに気付いています。プロセスアプローチでは、マネジメントシステムは経営ツールでもある為、組織ではなくプロセスという切り口で監査を行い、適切性、妥当性を判断し、経営者に報告、プロセスオーナーが改善を廻すというPDCAの枠組みが必要です。一方、現実はプロセスを監視し傾向管理を行っておらず、訓練もされていないにわか監査員が片手間に監査を行っているだけ。これは医療機器ビジネスでは致命的で、リコールで何十億の損失を出すだけではなく、人の命に関わる重大な問題に発展し、会社の存続まで危ぶまれます。このコースでは、ISO内部監査員の資格を持つ方々がQSRの内部監査が行えるよう社内資格を付与するための社内資格認定コースとして位置づけられます。一般に行われているISOの内部監査手法とは異なる、付加価値を生む予防対策としての監査手法とノウハウを学びます。( このコースを受講した後、オンサイトでクオリス・イノーバによる内部監査のOJTを行うとより効果的です )
日 時:2023年12月4日(月曜)、5日(火曜) 9:30 〜 16:30
会 場: 全国町村会館(東京・永田町) 永田町駅から徒歩1分( 3番出口 )
人数が少ない場合でも開催しますが、リモートのみになる可能性もございますので
予めご了承いただきますようお願いいたします。
対 象:すでに弊社主催下記コースをオープン、オンサイト問わず受講された方限定です。
* FDA GMP QSR 2日間コース
* FDA 設計管理2日間コース
このコースはQSRの基礎知識が無いと受講できず、弊社のコースを受講されていない場合
異なる解釈により内容をよく理解出来ません。 何卒ご理解をお願い申し上げます。
受講料: ¥99,000( 税込、テキスト・昼食含む )
割 引: ¥88,000( 税込、テキスト・昼食含む )
1社2名様以上受講される場合
定 員: 20名限定 満席になり次第締切ます
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
過去に受講された弊社QSR、設計管理各コースの受講日を記載して下さい。
オンサイトで受講された場合でも構いません。
折り返し、会場、お振込先情報をメールでご連絡致します。
セミナーに参加するための宿題を追って発送し致します。
2名様以上ご参加の場合、お名前とローマ字読み、
メールアドレスを連絡欄にご記入下さい。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.内部監査の目的を理解する
規制・規格が求めていること
患者の命を優先する監査とは
企業の付加価値を高める為には
2.内部監査員に求められる力量
内部監査員とリーダーに求められる力量
内部監査員訓練
マネジメントチームのカウンセラー
3.プロセスアプローチ
CAPAの理解とCAPAアプローチ
プロセスのR&R( 役割と責任 )を明確にする
プロセスを常に測定し改善する
内部監査はプロセスに注目
4.監査判定基準
チェックリストと数値化
プロセス毎に数値化
FDA査察と内部監査判断基準の相違を知る
5.監査を計画する
マネジメントレビューと品質会議からの指示
リスクベース&プロセスアプローチによる監査計画
プロセス管理パラメートと傾向分析結果
是正監査と予防監査の相違
監査は短期集中型で実施
監査員レビュー会のノウハウ
監査報告書記載のポイント
6.内部監査レポート
トップマネジメントに対する報告
プロセスの不足、改善点を報告
規制に対するコンプライアンス状況の報告
トップマネジメントのアクション
7.監査の目のつけどころ( QSIT )
QSITに基づく監査手法と勘所
プロセスアプローチの手法を全員が理解
1.経営者による管理
2.設計管理
3.CAPA
MDR報告
CAR
MDトラッキング
4.P&PC
5.滅菌工程
+6.各プロセス
*内容は変更される場合がございます*

2017_12_Internal auditor
購買プロセス リモートコース
〜 FDA 査察指摘 Worst 4 購買プロセスの本質と対策 〜
21CFR Part 820・ISO13485・ISO9001
このコースの売り上げ金の一部は人道支援活動に寄付されます
会場参加、リモート、ハイブリッド形式で開催されます
弊社コロナウィルス対策はこちらからご確認下さい
☆ 開催概要 ☆
FDA査察で必ず指摘を受ける(ワースト4)重要なプロセス。アッセンブラーが多くなった昨今、行政当局も購買プロセスに多くの査察時間を費やすようになりました。また、重要構成品のアウトソースを行う企業がほとんどで、実際はアウトソースとして扱うべきところを、パーツサプライヤとして扱ってみたり、マネジメントシステムの傘下として扱うべきサービス購買の要求事項を理解していません。さらに、購買プロセスは言われた通りにモノを買っていればよいというものではなく、サプライチェーン全体を俯瞰し、製品企画時の上流から積極的にコンカレントで参画、最も効率よいQCDを提案することこそ会社に貢献するプロセスとして価値があります。その為には、戦略的な組織体制とフレームを構築する必要があり、開発購買や情報購買と言ったフレームワークは早期の製品ローンチを実現します。新たな規制QMSRを想定し、ISO13485、9001の要求も加味しながら理解を深めます。
オンサイトでも可能です。お問い合わせはこちらから。
日 時:2023年11月20日(月曜) 9:30 〜 16:30
会 場:リモートのみ
( リモートでご参加なさる方は ZOOM でのご参加となります )
講 師:クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
ODM、OEMとして設計・製造を受託している企業
受講料: ¥55.000-( 消費税込、テキスト、ご来場の方には昼食を提供 )
リモートでご参加なさる方には、事前にテキスト等を郵送致します
割 引: ¥44.000-( 消費税込、テキスト、ご来場の方には昼食を提供 )
1社2名様以上ご参加の場合
特 割: 弊社プロジェクト実施中、継続教育等の場合ご相談下さい。
定 員: リモート 定員 40名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
申込締切: 11月13日(金曜)
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
![]()
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
A.FDA規制、査察の最新情報と傾向分析(QMSRの動向)
1.適用規制・規格
FDA QSR,ISO13485,9001の要求とは
プロセスアプローチの購買プロセスとは
購買プロセスの位置づけ
2.購買管理の目的
規格が示す購買管理の目的
製品、サービス購買とは
4つの購買範囲とは
3.用語の定義
製品購買、サービス購買とは
プロバイダー、サプライヤー、コントラクターとは
4.購買管理
GHTF( IMDRF )ガイダンスのフロー
5.購買方針
購買方針がなければ購買プロセスは成り立たない
6.戦略・開発購買
サプライチェーン全体を俯瞰した戦略
開発スピードを上げ原価削減、購買活動のスピードを上げある
ベストプラクティスの収集と技術の取り込み
7.購買計画
製品企画段階から参画
供給する製品、サービスの特定
技術及びプロセス情報の特定
供給者候補の特定
リスク、管理方法の特定
8.供給者候補選定
供給者ビジネス、運用能力評価
供給者候補選定
9.供給者選定
評価選定基準の策定
供給者候補の能力評価
供給者の承認
10.管理方法の取り決め
購買情報とは
管理方法
11.変更時対応
変更管理規定
4Mと変化点管理
12.納期、測定、監視
製品、サービスの検証
不適合の手順
供給者の監視
製品の受領、出荷場、冷蔵室、NCエリア
カーゴ内温度バリデーション
13.フィードバック、コミュニケーション
CR
SCAR
供給者評価シート
14.サプライヤー監査
リスクベース監査
5つの監査、評価エリア
情報購買、戦略上重要な監査チーム
( 内容が変更になる場合もございますが大きな変更はございません。)

2021_11_PC
FDA 設計管理2日間HBコース Rev.27
〜 リスクをヘッジしコンプライアンスを堅持するフレームワーク 〜
NEW QMSR 対応 21CFR Part 820.30 / 75 ISO13485
QSR内部監査員コースご出席要件コースです
( オンサイト及び過去弊社コースを受講された方も要件を満たします )
会場参加、リモート、ハイブリッド形式で開催します
☆ 開催概要 ☆
ほとんどの日本企業に於ける医療機器開発・設計管理の仕組みは世界標準にほど遠く、これはグローバルに進出しようとする企業のみならず、患者の命を預かる全ての医療機器メーカーにとって大きなリスクを伴います。米国FDAは自らの査察で、医療機器品質不良の根本原因は上流側の設計管理にあると査察の結果から統計的に見抜いており、それはクオリス・イノーバの15年以上にわたるGAP監査でも明らかです。そこで本コースでは、グローバルな開発、設計管理を米国FDA「設計管理ガイダンス」及び欧米で取り入れられている開発フレームワークを元に体系的に解説、また、品質不良を発生させない仕組み(ロバスト設計)をタグチメソッドと設計CAPAを用いて解説します。 2日目はプロセスバリデーションの本質、工程内試験に頼らない均一で高品質な製品を一貫して製造する為、パラメータによる製造方法を実現する工程設計について解説します。 ”規制” だからではなく、設計品質を高めることにより患者さんの命を守り、企業にとっても大きな潜在的利益を得られるという大原則は、エンジニアの方々から多くの共感と支持を頂いてきたクオリス・イノーバの最も人気のある価値あるコースです。 このコースはオンサイトでも実施しております。お問い合わせはこちらから。
日 時: 2023年11月6日(月曜)、7日(火曜) 9:30〜16:30
締 切: 2022年10月31日(火曜)
会 場: 全国町村会館 大ホール(東京・永田町) 永田町駅から徒歩1分( 3番出口 )
リモートで参加はZOOMとなります
講 師: クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象: 医療機器・IVD市場にこれから参入しようとしている企業
医療機器・IVDを海外に輸出しようとしている企業
FDA査察準備、MDSAPを受審しようと準備している企業
既にFDA査察を終え、指摘対応に苦慮している企業
製品実現に関わる各組織( マーケティング、開発、生産技術、品証など )
受講料: ¥99,000( 税込、テキスト・昼食含む )
割 引: ¥88,000( 税込、テキスト・昼食含む )
1社2名以上受講される場合
特 割: 再受講の方、プロジェクト終了企業様で継続研修としてご検討の方ご相談下さい。
定 員: クラス 定員 20名
リモート定員 30名
出席者全員に教育記録として利用できる修了書をお渡しします
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡いたします。
2名様以上ご参加の場合、お名前とローマ字読み、メールアドレスを連絡欄にご記入下さい。
持参品: 弊社QSR翻訳本第5版をご持参下さい。
お持ちでない方はこちらから特別価格でお求め頂けます。
ご注意: 本コースは、品質マネジメントシステムの一部を解説するため、システム全体の知識がないと理解できません。本コース受講前に10月開催QMSR2日間コースの受講をお勧めします。( QMSR2日間コースで訳本を配布しています )
開催要件:
1.コロナウイルス対応
弊社のウィルス対策を順守して戴きます。
当日熱がある場合、会場への出席はお控え頂きます。
2.リモートご参加要件
ZOOM以外での対応はしておりません
会社からご参加の場合、会議室から会社PCでのご参加となります。
自宅からご参加の場合、会社PCでのご参加となります。
その他、チェックリストでご確認頂く為、事前にご確認をお願いしています。
メールでお問い合わせ
電話でお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1日目(セミナー内容)
A.コンプライアンスとインテグリティー
設計不良の原因は何か
設計CAPAとは
ケーススタディ
不正を排除する仕組み
規制・規格に満足するればOKか?
人の命
行政によるミッションの相違
B.医療機器規制を理解する
新しい規則QMSRと今後の対応値
適用規制、規格、ガイダンス
1.Subpart C 設計管理 820.30 ISO13485 - 7.3
設計管理の目的とその重要性
プロセスアプローチの枠組と製品実現フレームワークの重要性
ライフサイクルマネジメント(LCM)と Waterfall Model
関連する ISO13485 の項目(プロセス)
Form 483 の FY 統計的解析から理解すること
2.820.30 (a) 総則 ISO13485 - 7.3.1
プロセスアプローチ ISO13485 4.1 の理解と
経営者の責任(リソースの提供とKPI)
製品実現プロセス
タートルチャート
リソースとしての力量(教育と訓練) 820.25
文書体系から俯瞰する設計管理プロセス
3.820.30 (b) 設計及び開発計画 ISO13485 - 7.3.2
医療機器開発ライフサイクルマネジメント(LCM)
設計品質確立の重要な要素; コンカレントエンジニアリング(CE)
PM、PLの役割と責任
ライフサイクルリスクマネジメントの理解(LCRM) ISO14971
タグチメソッド開発フレームワークによるフロントローディング手法
マーケティング戦略の重要性
開発計画書
4.820.30 (c) 設計インプット ISO13485 - 7.3.3
コミュニケーションエラー
曖昧なニーズの明確化
要求仕様から完成品までの一貫性
トレーサビリティーマトリックス(TMX)
ラベリング、包装設計のインプット
820.120 機器のラベリング
820.130 機器の包装
5.820.30 (d) 設計アウトプット ISO13485 - 7.3.4
タートルチャートのアウトプット
設計アウトプットのリスト
E−BOMの役割
製造仕様書とは
6.820.30 (e) 設計レビュー ISO13485 - 7.3.5
問題の早期発見、解決が目的でセレモニーで終わらせない仕組み
設計品質確立の重要な要素
設計レビューとテクニカルレビュー
レビューワーと責任と権限
7.820.30 (f) 設計検証 ISO13485 - 7.3.6
インプットからアウトプットまでのトレーサビリティー
製品の安全性、性能を証明するエビデンスの作り方
設計検証計画に必要な要素
設計検証レポートはデータの信頼性を確保
8.820.30 (g) 設計バリデーション ISO13485 - 7.3.7
設計検証と設計バリデーションの相違
設計バリデーションの手法
2つのバリデーションの相違と記録
ソフトウェアのバリデーションとVモデル
3種類のバリデーションの相違
9.820.30 (h) 設計移管 ISO13485 - 7.3.8 → 第2日目(工程設計)
10.820.30 (i ) 設計変更 ISO13485 - 7.3.9
変更管理プロセスに伴う設計変更
ライフサイクルリスクマネジメント
タートルチャート
設計変更フロー
11.820.30 (j ) DHF ISO13485 - 7.3.10 設計・開発ファイル
DHF / DMR / DHR の定義
識別、ファイルの基本
文書体系と設計標準
2日目(セミナー内容)
設計移管と工程設計
820.30 (h)、13485 - 7.3.8、7.5.6
1.820.30 (h) 設計移管 ISO13485 - 7.3.8
ライフサイクルリスクマネジメントにおける設計移管(工程設計)
均一な製品を一貫して生産する手法
プロセスバリデーションの目的
2.なぜ工程バリデーションを実施するのか 820.75, ISO13485 - 7.5.6
プロセスバリデーションの目的とその本質
プロセスバリデーションの効果
工程管理、監視の目的と本質
医療機器規制・規格の理解
3.バリデーションすべき工程とは
バリデーションすべき工程の識別
検証工程とバリデーション工程の相違
設備バリデーションと工程バリデーション
バリデーション工程例
4.バリデーションの種類
バリデーションの種類
設備バリデーションと劣化パラメータ 820.70(g), 72(a)
コンピューターシステムバリデーション(CSV) 820.70(i), ISO13485 - 7.5.6 (g)
5.バリデーションステップ 820.75, ISO13485 - 7.5.6
VMP:バリデーションマスタープラン
DQ: 設計時適格性評価
IQ: 据付時適格性評価
OQ: 稼働時適格性評価
PQ: 製造時適格性評価
PPQ: プロセス街道性評価
バリデーション文書化
プロセスバリデーションの明確化
自動化工程の明確化
CSVカテゴリーの明確化
ソフトウェアのバリデーションの基本
リスクベースCSV
6.工程設計とプロセスバリデーション 820.30 (h)、 ISO13485 - 7.3.8
ライフサイクルマネジメントに於ける工程設計
工程設計の進め方
工程設計とプロセスリスク分析( PFMEA )
環境管理 820.70(c)、 ISO13485 - 6.4
汚染管理 820.70(e)、 ISO13485 - 6.4
副資材 820.70(h)、 ISO13485 - 7.5.2
工程設計PTMX
QC工程表
7.回顧的バリデーション
8.バリデーションのアウトプット
QC工程表と統計的手法(工程能力)
リバリデーション設定
設備インスペクションとメンテナンス
( 内容が変更される場合がございます )

2023_05 設計管理2日間コース
〜 FDA査察指摘ワースト&キープロセスを理解する 〜
NEW QMSR 対応 21CFR Part 820.100、ISO13485 8.5、ISO9001 10
リモートコース
このコースの売上金の一部は人道支援に寄付されます
* コース概要 *
CAPAプロセスを確認するだけで、その会社の品質システムや品質に対する理解度がよくわかります。CAPAプロセスに問題があれば、品質マネジメントシステムの適切性、妥当性はなく、特にマネジメントレビューがまともに運用されているとは到底思われません。品質マネジメントシステムの原則はプロセスアプローチ、PDCAをまわす為、マネジメントレビューのインプットでプロセスを監視し、改善ツールであるCAPAでプロセスを改善します。当たり前のことがシステムとして運用されていないことを理解している規制当局は、CAPAプロセスにフォーカスして確認しようとします。QMSが機能していないことは昨今の品質に関わる事件から明らかで、医療機器・IVDの場合、命に関わる大問題です。本コースでは今までの内容を大きく変更し、プロセスアプローチの手法に重点を置き、事例を元にCAPAの本質を理解して頂きます。 このコースは受講者と同じ目線の御社事例を用いることでより理解が高まるオンサイトセミナーが可能です。お問合せはこちらから。
日 時:2023年10月20日(金曜)9:30 〜 16:30
会 場:ZOOMでリモートのみ開催
講 師:クオリス・イノーバ株式会社代表取締役社長 木村 浩実
対 象:医療機器、IVD、医薬品等に限定せず、CAPAプロセスを学びたい企業様
受講料: ¥55.000-( 税込、テキスト代 )
事前にテキストを郵送致します
割 引: ¥44.000-( 税込、テキスト代 )
1社2名様以上ご参加の場合
特 割: 弊社プロジェクト実施中、継続教育等の場合ご相談下さい
定 員: リモート 定員 30名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
開催要件:
1.リモート参加要件
会社からの場合、個室(会議室等)から会社PCで御参加となります。
ご自宅からの場合、会社PCでのご参加となります。
その他チェックリストを事前にお渡しし環境をご確認頂きます。
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.FDAがCAPAに注目している理由
警告書の分析
2.なぜ品質問題は再発するのか
直接原因と根本原因の相違
再発品質不良の根本原因
3.規制・規格を正しく理解する
CAPAの目的の理解
是正、予防、水平展開
統計的手法を使用し最初に予防
予防するシステムを理解する
4.TQMの仕組みが不可欠
トップがマネジメントするシステム
PDCAをまわす
プロセス改善ツール
5.リスクベースプロセスアプローチ
9001,13485CAPA
リスクベースプロセスアプローチとは?
測定、分析、改善
6.用語を正しく理解する
修正、是正、予防の相違
逸脱、不適合、逸脱許可と特別採用の相違
7.修正、是正、予防プロセス
受入アクセプタンス活動
工程内アクセプタンス活動
最終出荷アクセプタンス活動
苦情処理プロセス
設計変更プロセス
逸脱不適合プロセス
NCフローチャート
修理プロセス
8.CAPAプロセスエスカレーション
CA・PAエスカレーションの仕組み
リスク判断基準
9.CAPA7ステップ
Step 1 問題を検出し明確にする
Step 2 問題の原因を詳しく調査する
Step 3 根本原因の特定
Step 4 処置の特定と検証または妥当性確認
Step 5 予防または是正する為の処置
Step 6 処置の結果を文書化
Step 7 有効性の確認
10.フィードバック
フィードバックプロセス
CAPAはシステム改善プロセス
システムの有効性
11.品質問題の事例に学ぶ
Warning Letter Case Study
実際の事例

FDA QMSR/QSR/13485
2日間ハイブリッドコース
〜 医療機器・IVDに求められるMSの本質を学ぶ 〜
21CFR Part 820 / ISO13485 / ( ISO9001 )
このコースは会場、リモートのハイブリッド開催です。
2022年3月に公開された QSR に変わる新たな規制 QMSR について、
ISO13485 との相違はもとより、原点に立ち返る MS の本質について学ぶ新たなコースです。
年末に実施する監査員コースの受講要件となります。
☆ 開催概要 ☆
QSR翻訳本(第5版)+QMSR翻訳本(第1版)付き
米国が最も大きなマーケットであることは医療機器でも例外では無く、米国での成功がグローバルを制すると言っても過言ではありません。しかし、米国で製品を上市するには医療機器規制 QMSR に基づく QMS の構築は必須となります。2023年から新たに適用となるこの規制は、FDA QSR 規制を積極的に取り入れてきた ISO13485(2016) にほぼ準拠となりますが、ガイダンスなどは今まで通り適用となります。今後は QSR の欠点でもあった QMS の本質、リスクベースプロセスアプローチのフレームワークが必須となります。弊社の経験上 ISO1345 取得済みの企業であっても ISO に準拠しているとはとても言いがたい状況で QMSR には準拠できませんのでご注意下さい。今まで弊社は一貫して規制の本質を解き、事業活動とのインテグレーションを取ることにより、結果の出せるマネジメントシステムを提唱し、多くの企業の皆様の共感を集め支持されて参りました。米国に進出する企業のみならず、医療機器・IVDを開発しようとしている企業の皆様のマインドが大きく変わる価値あるクオリス・イノーバのメインコースです。
新規制が適用となるまで QSR が適用となる為、本コースでは現行 QSR, QSMR, ISO13485 について、リスクベースプロセスアプローチ、製品実現フレームワークといったシステムの根幹となるフレームワークを中心に概要を説明します。
オンサイト(一定人数が集まった会社でのセミナー)も可能です。お問合せはこちらから。
日 時: 2023年10月2日(月曜)、3日(火曜) 9:30〜16:30
会 場: 全国町村会館(東京・永田町)(永田町駅から徒歩1分)
リモートで参加なさる場合、ZOOMでの御参加となります。
講 師: クオリス・イノーバ株式会社
代表取締役社長 木村 浩実
対 象: 医療機器・IVD市場にこれから参入しようと考えている企業
米国に輸出を計画している企業
米国FDA 査察準備を考えている企業
ODM、OEM として医療機器を受託しようとしている企業
受講料: ¥99,000−(税込)( 2日分、テキスト、対訳本2冊、昼食代含 )
リモートで参加なさる方には事前にテキスト、対訳本を郵送いたします。
昼食は会場ご出席者のみ
割 引: ¥88,000−(税込)
1社2名以上ご参加の場合
特 割: 企業で行うQMS構築プロジェクトなどや企業で弊社が実施した継続研修などで
ご利用の場合、特別割引を適用させて頂きますのでメールでご相談下さい。
満席となった場合、正規料金をお支払いの方が優先となりご希望に添えない場合
がございますので予めご了承ください。
定 員: クラス 定員 20名様 9月26日締切
リモート定員 30名様
( 出席者全員に教育記録として利用できる修了書をお渡しします )
開催要件:
1.リモート参加要件
会社からの場合、個室(会議室等)から会社PCで御参加となります。
ご自宅からの場合、会社PCでのご参加となります。
その他チェックリストを事前にお渡しし環境をご確認頂きます。
2.COVID19対策
弊社のウィルス対策を順守して頂きます。
当日発熱がある場合、参加を御控え頂きます。
お申込: セミナー申込みページからお申し込み下さい。
折り返し、会場、お振り込み先情報などをメールでご案内致します。
2名以上で参加なさる方ははその旨ご記入下さい。
割引料金でご案内致します。
![]() メールでのお問い合わせ
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.コンプライアンスとインテグリティー
患者の命と会社の危機管理
データインテグリティー
2.医療機器規制の理解
行政によるミッションの相違
米国規制法
医療機器規制の理解
QSR vs QMSR
3.FDA査察
ISO企業の実態
FDA査察、頻度、査察タイプ
QSITアプローチ
A リスクベースプロセスアプローチ
Subpart_B 品質システム要求事項
820.20 経営者の責任
プロセスアプローチ
タートルチャート
マネジメントレビュー
文書体系
品質監査
力量
820.20 経営者の責任
Subpart_D 文書管理
820.40 文書管理
Subpart_O 統計的手法
820.250 統計的手法
Subpart_J 是正及び予防処置
820.100 CAPA
用語の正しい理解
エスカレーション
プロセスアプローチ
B 製品実現フレームワーク
Subpart_C 設計管理
Subpart_E 購買管理
Subpart_F 識別及びトレーサビリティー
Subpart_G 生産及び工程管理
Subpart_H アクセプランスアクティビティー
Subpart_I 不適合製品
Subpart_K ラベリング及び包装管理
Subpart_L 取扱い、保管、流通及び据付け
Subpart_M 記録
Subpart_N 附帯サービス
2023-05 QSR seminar

FDA MDR&CAR HBコース Rev.10
リモート参加も可能なハイブリッド形式
〜 査察指摘リスクが高い行政報告プロセス 〜
21CFR : Part 820, 803, 806
このコースの売り上げ金の一部は人道支援活動に寄付されます
☆ 開催概要 ☆
新しいQSRに変わるQMSRでISO13485にないプロセスを学び早期にプロセスを構築する必要があります。FDA査察レポートの解析からワースト4位(全体の約10%)に急浮上しているMDR(有害事象報告)、日本でも Warning Letter (警告書)を受けているほとんどの企業がMDRの指摘を受けています。昨今のデータ偽装事件を受け、FDAはこれらにフォーカスし厳しく査察を行っています。( 2021年レポート)。これはMDR報告の判断に迷ったら報告するという米国の文化とは異なり、日本のメーカーは情報を積極的に発信せず隠すという古い会社の文化が根強くあることが要因の一つですが、そもそも規制を理解せず正しい判断を行っていないことが大きな原因です。医療機器を作っているメーカーは、患者さんの命を守るという社会的な責任があります。会社を守っているようでも、結果的に会社が倒産に追い込まれることもある非常にリスクの高いプロセスであることを理解すべきです。それには規制の正しい理解、規制を守る仕組みと組織が必要です。このセミナーでは、CAPAサブシステム(苦情、不適合、CAPA)の各プロセスとリンクし、日本のメーカーの Warning Letter (警告書)を引き合いに正しいMDRプロセスと関連するCAR(リコール)プロセスを構築することを理解します。
このコースはオンサイトでも実施しています。お問い合わせはこちらから。
日 時:2023年9月11日(月曜)10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
リモートはZOOMから
対 象:既に医療機器・IVDを米国に輸出している企業
これから医療機器・IVDを米国輸出する準備を行っている企業
既にFDA査察を終え対応に困っている企業
受講料: ¥55,000-( 消費税込み、テキスト代含む、ご来場の方には昼食代含まれます )
リモートの方には事前にテキストを郵送させて頂きます
割 引: ¥44,000-( 消費税込み、テキスト代含む、ご来場の方には昼食代含まれます )
1社2名以上ご出席の場合
定 員: 20名( 会場 )リモート(30名)
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
特 割:弊社プロジェクトを実施中、又は継続研修をご希望の場合、御負担軽減のためさらに
割引をさせて頂きますのでお問い合わせください。この場合、正規料金でお申込の方
が優先され、出席できない場合がございますので予めご了承ください。
再受講:再受講をご希望の方はさらに割引が適用され、空席がある場合ご出席頂けます。
併せて過去ご出席なさった日をお知らせ下さい。
お申込:セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
リモートをご希望の方はその旨ご記入下さい。
開催要件:リモート参加の場合、リモート環境に確認とNDAへのサインをお願いしています。
パンデミックによる行政指導がある場合、ご来場の方もリモートに切り替えて頂き、
開催となります。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.規制を理解する
過去事例と統計からみる規制違反
理解すべき規制・規格・ガイダンス
eMDR規制
苦情組織と力量
規制が求めるあるべき組織と力量
プロセス構成
2.有害事象報告( MDR )Part 803
MDR規制の基本要件
FDA Warning letter から
規制の目的、重要性
MDRとして報告すべき事象とは
2.1 定義を理解する
重篤な障害
機能不全
報告すべき Malfunction
合理的に推測する
認識する
2.2 4.5日、30日報告
報告形態と時間軸
5日報告
ディシジョンツリー
2.3 FDAへの報告
報告フロー
報告様式 3500A
報告不要の場合
報告書の種類
報告免除
Warning letter 警告書
2.4 MDRプロセス
MDR報告フロー
CA,PAプロセス
3.回収報告(CAR)Part 806
規制の理解
報告免除
定義
リコールフロー
任意と強制リコール
定期メンテナンスとリコール
3.1 リコール判断基準
リコール判断基準例
ラベリングの定義
ラベリング違反と法的根拠
報告すべきラベリング違反例
3.2 CAR ディシジョンツリー
リコールディシジョンツリー
3.3 報告書と処理フロー
リコール判断後フロー
リコールクラス
有効性の確認
CAPAとの関係
4.警告書から理解すべき注意点とプロセスへの展開
Warning Letter ( 警告書 )の事例から理解する
警告書から理解すべき注意点とプロセスへの展開
プロセス構築後のレトロスペクティブな是正とは
*コース内容が一部変更される場合がございますが大きな変更はございません。
〜 FDA査察指摘ワースト&キープロセス 〜
NEW QMSR 対応 21CFR Part 820.100、ISO13485 8.5
会場、リモート対応のハイブリッドコース!
このコースの売上金の一部は人道支援に寄付されます
* コース概要 *
CAPAプロセスを確認するだけで、その会社の品質システムや品質に対する理解度がよくわかります。CAPAプロセスに問題があれば、品質マネジメントシステムの適切性、妥当性はなく、特にマネジメントレビューがまともに運用されているとは到底思われません。品質マネジメントシステムの原則はプロセスアプローチ、PDCAをまわす為、マネジメントレビューのインプットでプロセスを監視し、改善ツールであるCAPAでプロセスを改善します。当たり前のことがシステムとして運用されていないことを理解している規制当局は、CAPAプロセスにフォーカスして確認しようとします。QMSが機能していないことは昨今の品質に関わる事件から明らかで、医療機器・IVDの場合、命に関わる大問題です。本コースでは今までの内容を大きく変更し、プロセスアプローチの手法に重点を置き、事例を元にCAPAの本質を理解して頂きます。 このコースは受講者と同じ目線の御社事例を用いることでより理解が高まるオンサイトセミナーが可能です。お問合せはこちらから。
日 時:2023年7月3日(月曜)9:30 〜 16:30
会 場:全国町村会館( 東京・永田町 ) ( 永田町駅から徒歩1分 )
リモート参加はZOOMで行います
講 師:クオリス・イノーバ株式会社代表取締役社長 木村 浩実
対 象:医療機器、IVD、医薬品等に限定せず、CAPAプロセスを学びたい企業様
受講料: ¥55.000-( 消費税込、テキスト、ご来場の方には昼食を提供 )
リモートでご参加なさる方には、事前にテキスト等を郵送致します
割 引: ¥44.000-( 消費税込、テキスト、ご来場の方には昼食を提供 )
1社2名様以上ご参加の場合
特 割: 弊社プロジェクト実施中、継続教育等の場合ご相談下さい。
定 員: クラス 定員 20名
リモート定員 30名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
開催要件:
1.リモート参加要件
会社からの場合、個室(会議室等)から会社PCで御参加となります。
ご自宅からの場合、会社PCでのご参加となります。
その他チェックリストを事前にお渡しし環境をご確認頂きます。
2.コロナウィルス対応
弊社のウィルス対策を順守して頂きます。
当日発熱がある場合、参加をお控え頂きます。
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.なぜFDAはCAPAに注目するのか
警告書の分析
2.なぜ品質問題は再発するのか
3.規制・規格を正しく理解する
QMSR/ISO
4.TQMの仕組みが不可欠
トップマネジメント
タートルチャート
CAPAマネジメント
5.リスクベースプロセスアプローチ
経営の本質
プロセスの廻し方
5.用語を正しく理解する
修正、是正、予防の相違
逸脱、不適合、逸脱許可と特別採用の相違
修正、手直し、修理の相違
7.修正、是正、予防プロセス
アクセプタンスアクティビティー
苦情処理プロセス
設計変更プロセス
逸脱・不適合処理プロセス
修理プロセス
8.CAPAエスカレーション
CA/PA 解析データソース
CA/PA エスカレーション
PA エスカレーション
エスカレーションレベル
CA エスカレーション
9.CAPA7ステップ
PA詳細フロー1(直接原因)
PA詳細フロー2(根本原因)
CA詳細フロー1(直接原因)
CA詳細フロー2(根本原因)
CAPA7ステップ
10.フィードバック
フィードバックプロセス
システム改善ツール
11.品質問題の事例に学ぶ
Warninng Letter 4
Warninng Letter 5
Warninng Letter 6
例示1
例示2
Case Study 1
Case Study 2
Case Study 3
* 内容(項目)が変更される場合があります
2022_7_CAPA course
QMS 購買プロセスHBコース
〜 FDA 査察指摘 Worst 4 購買プロセスの本質と対策 〜
21CFR Part 820・ISO13485・ISO9001
このコースの売り上げ金の一部は人道支援活動に寄付されます
会場参加、リモート、ハイブリッド形式で開催されます
弊社コロナウィルス対策はこちらからご確認下さい
☆ 開催概要 ☆
購買プロセスは言われた通りにモノを買っていればよいというものではなく、サプライチェーン全体を俯瞰し、製品企画時の上流から積極的にコンカレントで参画、最も効率よいQCDを提案することこそ会社に貢献するプロセスとして価値があります。その為には、戦略的な組織体制とフレームを構築する必要があり、開発購買や情報購買と言ったフレームワークは早期の製品ローンチを実現します。また、アッセンブルラーが多くなった昨今、行政当局も購買プロセスに多くの査察時間を費やすようになりました。自社と同じシステムで管理する必要性が高まり、フレームと管理能力が要求されます。FDA QSRのみならず、ISOの基本9001の要求も加味しながら理解を深めます。
オンサイトでも可能です。お問い合わせはこちらから。
日 時:2023年6月19日(月曜) 9:30 〜 16:30
会 場:全国町村会館(東京・永田町) 永田町駅から徒歩1分( 3番出口 )
リモートでご参加なさる方は ZOOM でのご参加となります
講 師:クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
ODM、OEMとして設計・製造を受託している企業
受講料: ¥55.000-( 消費税込、テキスト、ご来場の方には昼食を提供 )
リモートでご参加なさる方には、事前にテキスト等を郵送致します
割 引: ¥44.000-( 消費税込、テキスト、ご来場の方には昼食を提供 )
1社2名様以上ご参加の場合
特 割: 弊社プロジェクト実施中、継続教育等の場合ご相談下さい。
定 員: クラス 定員 20名
リモート定員 30名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
開催要件:
1.リモート参加要件
会社からの場合、個室(会議室等)から会社PCで御参加となります。
ご自宅からの場合、会社PCでのご参加となります。
その他チェックリストを事前にお渡しし環境をご確認頂きます。
2.コロナウィルス対応
弊社のウィルス対策を順守して頂きます。
当日発熱がある場合、参加を御控え頂きます。
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
A.FDA規制、査察の最新情報と傾向分析(QMSRの動向)
1.適用規制・規格
FDA QSR,ISO13485,9001の要求とは
プロセスアプローチの購買プロセスとは
購買プロセスの位置づけ
2.購買管理の目的
規格が示す購買管理の目的
製品、サービス購買とは
4つの購買範囲とは
3.用語の定義
製品購買、サービス購買とは
プロバイダー、サプライヤー、コントラクターとは
4.購買管理
GHTF( IMDRF )ガイダンスのフロー
5.購買方針
購買方針がなければ購買プロセスは成り立たない
6.戦略・開発購買
サプライチェーン全体を俯瞰した戦略
開発スピードを上げ原価削減、購買活動のスピードを上げある
ベストプラクティスの収集と技術の取り込み
7.購買計画
製品企画段階から参画
供給する製品、サービスの特定
技術及びプロセス情報の特定
供給者候補の特定
リスク、管理方法の特定
8.供給者候補選定
供給者ビジネス、運用能力評価
供給者候補選定
9.供給者選定
評価選定基準の策定
供給者候補の能力評価
供給者の承認
10.管理方法の取り決め
購買情報とは
管理方法
11.変更時対応
変更管理規定
4Mと変化点管理
12.納期、測定、監視
製品、サービスの検証
不適合の手順
供給者の監視
製品の受領、出荷場、冷蔵室、NCエリア
カーゴ内温度バリデーション
13.フィードバック、コミュニケーション
CR
SCAR
供給者評価シート
14.サプライヤー監査
リスクベース監査
5つの監査、評価エリア
情報購買、戦略上重要な監査チーム
( 内容が変更になる場合もございますが大きな変更はございません。)
2021_11_PC
FDA 設計管理2日間HBコース Rev.27
〜 リスクをヘッジしコンプライアンスを堅持するフレームワーク 〜
NEW QMSR 対応 21CFR Part 820.30 / 75 ISO13485
QSR内部監査員コースご出席要件コースです
( オンサイト及び過去弊社コースを受講された方は要件を満たします )
会場参加、リモート、ハイブリッド形式で開催します
☆ 開催概要 ☆
ほとんどの日本企業に於ける医療機器開発・設計管理の仕組みは世界標準にほど遠く、これはグローバルに進出しようとする企業のみならず、患者の命を預かる全ての医療機器メーカーにとって大きなリスクを伴います。米国FDAは自らの査察で、医療機器品質不良の根本原因は上流側の設計管理にあると査察の結果から統計的に見抜いており、それはクオリス・イノーバの10年以上にわたるGAP監査でも明らかです。そこで本コースでは、グローバルな開発、設計管理を米国FDA「設計管理ガイダンス」及び欧米で取り入れられている開発フレームワークを元に体系的に解説、また、品質不良を発生させない仕組み(ロバスト設計)をタグチメソッドと設計CAPAを用いて解説します。 2日目はプロセスバリデーションの本質、工程内試験に頼らない均一で高品質な製品を一貫して製造する為、パラメータによる製造方法を実現する工程設計について解説します。 ”規制” だからではなく、設計品質を高めることにより患者さんの命を守り、企業にとっても大きな潜在的利益を得られるという大原則は、エンジニアの方々から多くの共感と支持を頂いてきたクオリス・イノーバの最も人気のある価値あるコースです。 このコースはオンサイトでも実施しております。お問い合わせはこちらから。
日 時: 2023年6月5日(月曜)、6日(火曜) 9:30〜16:30
締 切: 2022年5月30日(火曜)
会 場: 全国町村会館 大ホール(東京・永田町) 永田町駅から徒歩1分( 3番出口 )
リモートで参加はZOOMとなります
講 師: クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象: 医療機器・IVD市場にこれから参入しようとしている企業
医療機器・IVDを海外に輸出しようとしている企業
FDA査察準備、MDSAPを受審しようと準備している企業
既にFDA査察を終え、指摘対応に苦慮している企業
製品実現に関わる各組織( マーケティング、開発、生産技術、品証など )
受講料: ¥99,000( 税込、テキスト・昼食含む )
割 引: ¥88,000( 税込、テキスト・昼食含む )
1社2名様以上受講される場合
特 割: 再受講の方、プロジェクト終了企業様で継続研修としてご検討の方ご相談下さい。
定 員: クラス 定員 20名
リモート定員 30名
出席者全員に教育記録として利用できる修了書をお渡しします
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡いたします。
2名様以上ご参加の場合、お名前とローマ字読み、メールアドレスを連絡欄にご記入下さい。
持参品: 弊社QSR翻訳本第5版をご持参下さい。
お持ちでない方はこちらから特別価格でお求め頂けます。
ご注意: 本コースは、品質マネジメントシステムの一部を解説するため、システム全体の知識がないと理解できません。本コース受講前に5月開催QMSR2日間コースの受講をお勧めします。( QMSR2日間コースで訳本を配布しています )
開催要件:
1.コロナウイルス対応
行政の指示による規制の対象となった場合、リモートのみの開催となります。
弊社のウィルス対策を順守して戴きます。
2.リモートご参加要件
ZOOM以外での対応はしておりません
会社からご参加の場合、会議室から会社PCでのご参加となります。
自宅からご参加の場合、会社PCでのご参加となります。
その他、チェックリストでご確認頂く為、事前にご確認をお願いしています。
メールでお問い合わせ
電話でお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1日目(セミナー内容)
A.コンプライアンスとインテグリティー
設計不良の原因は何か
設計CAPAとは
ケーススタディ
不正を排除する仕組み
規制・規格に満足するればOKか?
人の命
行政によるミッションの相違
B.医療機器規制を理解する
新しい規則QMSRと今後の対応値
適用規制、規格、ガイダンス
1.Subpart C 設計管理 820.30 ISO13485 - 7.3
設計管理の目的とその重要性
プロセスアプローチの枠組と製品実現フレームワークの重要性
ライフサイクルマネジメント(LCM)と Waterfall Model
関連する ISO13485 の項目(プロセス)
Form 483 の FY 統計的解析から理解すること
2.820.30 (a) 総則 ISO13485 - 7.3.1
プロセスアプローチ ISO13485 4.1 の理解と
経営者の責任(リソースの提供とKPI)
製品実現プロセス
タートルチャート
リソースとしての力量(教育と訓練) 820.25
文書体系から俯瞰する設計管理プロセス
3.820.30 (b) 設計及び開発計画 ISO13485 - 7.3.2
医療機器開発ライフサイクルマネジメント(LCM)
設計品質確立の重要な要素; コンカレントエンジニアリング(CE)
PM、PLの役割と責任
ライフサイクルリスクマネジメントの理解(LCRM) ISO14971
タグチメソッド開発フレームワークによるフロントローディング手法
マーケティング戦略の重要性
開発計画書
4.820.30 (c) 設計インプット ISO13485 - 7.3.3
コミュニケーションエラー
曖昧なニーズの明確化
要求仕様から完成品までの一貫性
トレーサビリティーマトリックス(TMX)
ラベリング、包装設計のインプット
5.820.30 (d) 設計アウトプット ISO13485 - 7.3.4
タートルチャートのアウトプット
設計アウトプットのリスト
E−BOMの役割
製造仕様書とは
6.820.30 (e) 設計レビュー ISO13485 - 7.3.5
問題の早期発見、解決が目的でセレモニーで終わらせない仕組み
設計品質確立の重要な要素
設計レビューとテクニカルレビュー
レビューワーと責任と権限
7.820.30 (f) 設計検証 ISO13485 - 7.3.6
インプットからアウトプットまでのトレーサビリティー
製品の安全性、性能を証明するエビデンスの作り方
設計検証計画に必要な要素
設計検証レポートはデータの信頼性を確保
8.820.30 (g) 設計バリデーション ISO13485 - 7.3.7
設計検証と設計バリデーションの相違
設計バリデーションの手法
2つのバリデーションの相違と記録
ソフトウェアのバリデーションとVモデル
3種類のバリデーションの相違
9.820.30 (h) 設計移管 ISO13485 - 7.3.8 → 第2日目(工程設計)
10.820.30 (i ) 設計変更 ISO13485 - 7.3.9
変更管理プロセスに伴う設計変更
ライフサイクルリスクマネジメント
タートルチャート
設計変更フロー
11.820.30 (i ) 設計変更 ISO13485 - 7.3.10 設計・開発ファイル
DHF / DMR / DHR の定義
識別、ファイルの基本
文書体系と設計標準
2日目(セミナー内容)
設計移管と工程設計
820.30 (h)、13485 - 7.3.8、7.5.6
1.820.30 (h) 設計移管 ISO13485 - 7.3.8
ライフサイクルリスクマネジメントにおける設計移管(工程設計)
均一な製品を一貫して生産する手法
プロセスバリデーションの目的
2.なぜ工程バリデーションを実施するのか 820.75, ISO13485 - 7.5.6
プロセスバリデーションの目的とその本質
プロセスバリデーションの効果
工程管理、監視の目的と本質
医療機器規制・規格の理解
3.バリデーションすべき工程とは
バリデーションすべき工程の識別
検証工程とバリデーション工程の相違
設備バリデーションと工程バリデーション
バリデーション工程例
4.バリデーションの種類
バリデーションの種類
設備バリデーションと劣化パラメータ 820.70(g), 72(a)
コンピューターシステムバリデーション(CSV) 820.70(i), ISO13485 - 7.5.6 (g)
5.バリデーションステップ 820.75, ISO13485 - 7.5.6
VMP:バリデーションマスタープラン
DQ: 設計時適格性評価
IQ: 据付時適格性評価
OQ: 稼働時適格性評価
PQ: 製造時適格性評価
PPQ: プロセス街道性評価
バリデーション文書化
プロセスバリデーションの明確化
自動化工程の明確化
CSVカテゴリーの明確化
ソフトウェアのバリデーションの基本
リスクベースCSV
6.工程設計とプロセスバリデーション 820.30 (h)、 ISO13485 - 7.3.8
ライフサイクルマネジメントに於ける工程設計
工程設計の進め方
工程設計とプロセスリスク分析( PFMEA )
環境管理 820.70(c)、 ISO13485 - 6.4
汚染管理 820.70(e)、 ISO13485 - 6.4
副資材 820.70(h)、 ISO13485 - 7.5.2
工程設計PTMX
QC工程表
7.回顧的バリデーション
8.バリデーションのアウトプット
QC工程表と統計的手法(工程能力)
リバリデーション設定
設備インスペクションとメンテナンス
( 内容が変更される場合がございます )

2022_06 設計管理2日間コース
FDA QMSR/QSR/13485
2日間HBコース
〜 医療機器・IVDに求められるQMSの本質を学ぶ 〜
21CFR Part 820 / ISO13485
このコースは会場、リモートのハイブリッド開催です。
2022年3月に公開された QSR に変わる新たな規制 QMSR について、
ISO13485 との相違はもとより、原点に立ち返る QMS の本質について学ぶ新たなコースです。
年末に実施する監査員コースの受講要件となります。
☆ 開催概要 ☆
QSR翻訳本(第5版)+QMSR翻訳本(第1版)付き
米国が最も大きなマーケットであることは医療機器でも例外では無く、米国での成功がグローバルを制すると言っても過言ではありません。しかし、米国で製品を上市するには医療機器規制 QMSR に基づく QMS の構築は必須となります。2023年から新たに適用となるこの規制は、FDA QSR 規制を積極的に取り入れてきた ISO13485(2016) にほぼ準拠となりますが、ガイダンスなどは今まで通り適用となります。今後は QSR の欠点でもあった QMS の本質、リスクベースプロセスアプローチのフレームワークが必須となります。弊社の経験上 ISO1345 取得済みの企業であっても ISO に準拠しているとはとても言いがたい状況で QMSR には準拠できませんのでご注意下さい。今まで弊社は一貫して規制の本質を解き、事業活動とのインテグレーションを取ることにより、結果の出せるマネジメントシステムを提唱し、多くの企業の皆様の共感を集め支持されて参りました。米国に進出する企業のみならず、医療機器・IVDを開発しようとしている企業の皆様のマインドが大きく変わる価値あるクオリス・イノーバのメインコースです。
新規制が適用となるまで QSR が適用となる為、本コースでは現行 QSR, QSMR, ISO13485 について、リスクベースプロセスアプローチ、製品実現フレームワークといったシステムの根幹となるフレームワークを中心に概要を説明します。
オンサイト(一定人数が集まった会社でのセミナー)も可能です。お問合せはこちらから。
日 時: 2023年5月22日(月曜)、23日(火曜) 9:30〜16:30
会 場: 全国町村会館(東京・永田町)(永田町駅から徒歩1分)
リモートで参加なさる場合、ZOOMでの御参加となります。
講 師: クオリス・イノーバ株式会社
取締役社長 木村 浩実
対 象: 医療機器・IVD市場にこれから参入しようと考えている企業
米国に輸出を計画している企業
米国FDA 査察準備を考えている企業
ODM、OEM として医療機器を受託しようとしている企業
受講料: ¥99,000−(税込)( 2日分、テキスト、対訳本2冊、昼食代含 )
リモートで参加なさる方には事前にテキスト、対訳本を郵送いたします。
昼食は会場ご出席者のみ
割 引: ¥88,000−(税込)
1社2名以上ご参加の場合
特 割: 企業で行うQMS構築プロジェクトなどや企業で弊社が実施した継続研修などで
ご利用の場合、特別割引を適用させて頂きますのでメールでご相談下さい。
満席となった場合、正規料金をお支払いの方が優先となりご希望に添えない場合
がございますので予めご了承ください。
定 員: クラス 定員 20名様 5月16日締切
リモート定員 30名様
( 出席者全員に教育記録として利用できる修了書をお渡しします )
開催要件:
1.リモート参加要件
会社からの場合、個室(会議室等)から会社PCで御参加となります。
ご自宅からの場合、会社PCでのご参加となります。
その他チェックリストを事前にお渡しし環境をご確認頂きます。
2.COVID19対策
弊社のウィルス対策を順守して頂きます。
当日発熱がある場合、参加を御控え頂きます。
お申込: セミナー申込みページからお申し込み下さい。
折り返し、会場、お振り込み先情報などをメールでご案内致します。
2名以上で参加なさる方ははその旨ご記入下さい。
割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.コンプライアンスとインテグリティー
患者の命と会社の危機管理
データインテグリティー
2.医療機器規制の理解
行政によるミッションの相違
米国規制法
医療機器規制の理解
3.QSR から QMSR へ移行する経緯
4.QSR と QMSR 、ISO13485( 2016) との相違
5.QMSR 移行後変わるものと変わらないもの
6.なぜ ISO 認証では FDA 査察にパスしないのか
7.QSIT アプローチとは
8.QMSR
Subpart A 総則
820.1 適用範囲
820.3 定義
規制を正しく理解する要素:定義
820.7 参照による引用
規制に記載していない ISO の引用について
820.10 品質マネジメントシステムに関する要求事項
ISO13485(2016) 適用の要件
820.15 コンセプトの明確化
Subpart B 附則
820.35 記録の管理
820.45 機器のラベリング及び包装管理
9.QMS を構成するフレームワーク
QSR ISO13485 (2016)
A. リスクベースプロセスアプローチ
4. 品質マネジメントシステム
820.20 経営者の責任 5. 経営者の責任
820.22 品質監査 8.2.4 内部監査
820.25 要員 6. 資源の運用管理
820.40 文書管理 4.2 文書化に関する要求事項
820.90 不適合製品 8.3 不適合製品の管理
820.100 是正処置及び予防処置 8.5 改善
820.250 統計的手法 8 測定、分析及び改善
B.製品実現フレームワーク
820.30 設計管理 7.3 設計・開発
820.50 購買管理 7.4 購買
820.60 識別 7.5.8 識別
820.65 トレーサビリティー 7.5.9 トレーサビリティー
820.70 生産及びプロセスの管理 7.5 製造及びサービスの提供
820.72 検査、測定及び検査装置 7.6 監視機器及び測定機器の管理
820.75 プロセスバリデーション 7.5.6 プロセスのバリデーション
820.80 受入、工程内及び完成機器のアクセプタンス
7.4.3 購買製品の検証
820.86 アクセプタンス状態 7.5.8 識別
820.120 機器のラベリング
820.130 機器の包装
820.140 取扱い 7.5.11 製品の保存
820.150 保管 7.5.11 製品の保存
820.160 流通 7.5.11 製品の保存
820.170 据付け 7.5.3 据付け活動
820.180 (記録) 4.2.5 記録の管理
820.181 DMR 4.2.3 医療機器ファイル
820.184 DHR 7.1 製品実現の計画
820.186 QSR 4.2.4 文書管理
820.198 苦情ファイル 8.2.2 苦情ファイル
820.200 附帯サービス 7.5.4 附帯サービス活動
2022-05 QSR seminar

〜 FDA査察指摘ワースト&キープロセスを理解する 〜
NEW QMSR 対応 21CFR Part 820.100、ISO13485 8.5
リモートコース!
このコースの売上金の一部は人道支援に寄付されます
* コース概要 *
CAPAプロセスを確認するだけで、その会社の品質システムや品質に対する理解度がよくわかります。CAPAプロセスに問題があれば、品質マネジメントシステムの適切性、妥当性はなく、特にマネジメントレビューがまともに運用されているとは到底思われません。品質マネジメントシステムの原則はプロセスアプローチ、PDCAをまわす為、マネジメントレビューのインプットでプロセスを監視し、改善ツールであるCAPAでプロセスを改善します。当たり前のことがシステムとして運用されていないことを理解している規制当局は、CAPAプロセスにフォーカスして確認しようとします。QMSが機能していないことは昨今の品質に関わる事件から明らかで、医療機器・IVDの場合、命に関わる大問題です。本コースでは今までの内容を大きく変更し、プロセスアプローチの手法に重点を置き、事例を元にCAPAの本質を理解して頂きます。 このコースは受講者と同じ目線の御社事例を用いることでより理解が高まるオンサイトセミナーが可能です。お問合せはこちらから。
日 時:2023年4月21日(金曜)9:30 〜 16:30
会 場:ZOOMでリモート開催
講 師:クオリス・イノーバ株式会社代表取締役社長 木村 浩実
対 象:医療機器、IVD、医薬品等に限定せず、CAPAプロセスを学びたい企業様
受講料: ¥55.000-( 税込、テキスト代 )
事前にテキストを郵送致します
割 引: ¥44.000-( 税込、テキスト代 )
1社2名様以上ご参加の場合
特 割: 弊社プロジェクト実施中、継続教育等の場合ご相談下さい。
定 員: リモート 定員 30名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
開催要件:
1.リモート参加要件
会社からの場合、個室(会議室等)から会社PCで御参加となります。
ご自宅からの場合、会社PCでのご参加となります。
その他チェックリストを事前にお渡しし環境をご確認頂きます。
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.なぜFDAはCAPAに注目するのか
警告書の分析
2.なぜ品質問題は再発するのか
3.規制・規格を正しく理解する
QMSR/ISO
4.プロセスアプローチの基本
プロセス関連図
タートルチャート
5.TQMの仕組みが不可欠
マネジメントレビュー
PDCAをまわす
プロセス改善ツール
6.用語の正しい理解
修正、是正、予防の相違
逸脱、不適合、逸脱許可と特別採用の相違
修正、手直し、修理の相違
7.CAPAプロセスの IN・OUT
CAPAタートルチャート
不適合処理プロセス
苦情処理プロセス
CAPAプロセス
MDRプロセス
リコールプロセス
エスカレーションの仕組み
8.直接原因と根本原因を理解する
直接原因と根本原因の相違
根本原因の改善がプロセスアプローチの本質
9.予防プロセス
品質管理の原則、予防活動の理解
10.エスカレーション
CAPAエスカレーション
CAPAを上手に運用するための仕組み
11.CAPA7ステップ
Step 1 問題を検出し明確にする
Step 2 問題の原因を詳しく調査する
Step 3 根本原因の特定
Step 4 処置の特定と検証または妥当性確認
Step 5 予防または是正する為の処置
Step 6 処置の結果を文書化
Step 7 有効性の確認
12.SCARの実施
13.設計CAPA
14.CAPAマネジメント
マネジメントレビューの運用方法
15.品質問題の事例に学ぶ
Warning Letter Case Study
実際の事例
FDA QMSR/QSR/13485 1日リモートコース
〜 医療機器・IVDに求められるQMSの本質を学ぶ 〜
21CFR Part 820
このコースは、リモートのみの開催です。
初めてQSRを学ぶ方や忙しい方向けのQSRの概要説明コースです
☆ 開催概要 ☆
QSR翻訳本(第5版)付き
米国が最も大きなマーケットであることは医療機器でも例外では無く、米国での成功がグローバルを制すると言っても過言ではありません。しかし、米国で製品を上市するには医療機器規制 QMSR に基づく QMS の構築は必須となります。2023年から新たに適用となるこの規制は、FDA QSR 規制を積極的に取り入れてきた ISO13485(2016) にほぼ準拠となりますが、ガイダンスなどは今まで通り適用となります。今後は QSR の欠点でもあった QMS の本質、リスクベースプロセスアプローチのフレームワークが必須となります。弊社の経験上 ISO1345 取得済みの企業であっても ISO に準拠しているとはとても言いがたい状況で QMSR には準拠できませんのでご注意下さい。今まで弊社は一貫して規制の本質を解き、事業活動とのインテグレーションを取ることにより、結果の出せるマネジメントシステムを提唱し、多くの企業の皆様の共感を集め支持されて参りました。米国に進出する企業のみならず、医療機器・IVDを開発しようとしている企業の皆様のマインドが大きく変わる価値あるクオリス・イノーバのメインコースの1日バージョンです。
オンサイト(一定人数が集まった会社でのセミナー)も可能です。お問合せはこちらから。
日 時: 2023年4月24日(月曜) 9:30〜16:30
会 場: ZOOMでリモート開催
講 師: クオリス・イノーバ株式会社
取締役社長 木村 浩実
対 象: 医療機器・IVD市場にこれから参入しようと考えている企業
米国に輸出を計画している企業
米国FDA 査察準備を考えている企業
ODM、OEM として医療機器を受託しようとしている企業
受講料: ¥55,000−(税込)( テキスト、対訳本1冊 )
事前にテキスト、対訳本を郵送いたします。
割 引: ¥44,000−(税込)
1社2名以上ご参加の場合
特 割: 企業で行うQMS構築プロジェクトなどや企業で弊社が実施した継続研修などで
ご利用の場合、特別割引を適用させて頂きますのでメールでご相談下さい。
満席となった場合、正規料金をお支払いの方が優先となりご希望に添えない場合
がございますので予めご了承ください。
定 員: リモート 定員 30名様 4月17日締切
( 出席者全員に教育記録として利用できる修了書をお渡しします )
開催要件:
1.リモート参加要件
会社からの場合、個室(会議室等)から会社PCで御参加となります。
ご自宅からの場合、会社PCでのご参加となります。
その他チェックリストを事前にお渡しし環境をご確認頂きます。
お申込: セミナー申込みページからお申し込み下さい。
折り返し、会場、お振り込み先情報などをメールでご案内致します。
2名以上で参加なさる方ははその旨ご記入下さい。
割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
項目は2日間コースと変わりませんが、内容が簡易的な説明となります
1.コンプライアンスとインテグリティー
患者の命と会社の危機管理
データインテグリティー
2.医療機器規制の理解
行政によるミッションの相違
米国規制法
医療機器規制の理解
3.QSR から QMSR へ移行する経緯
4.QSR と QMSR 、ISO13485( 2016) との相違
5.QMSR 移行後変わるものと変わらないもの
6.なぜ ISO 認証では FDA 査察にパスしないのか
7.QSIT アプローチとは
8.QMSR
Subpart A 総則
820.1 適用範囲
820.3 定義
規制を正しく理解する要素:定義
820.7 参照による引用
規制に記載していない ISO の引用について
820.10 品質マネジメントシステムに関する要求事項
ISO13485(2016) 適用の要件
820.15 コンセプトの明確化
Subpart B 附則
820.35 記録の管理
820.45 機器のラベリング及び包装管理
9.QMS を構成するフレームワーク
QSR ISO13485 (2016)
A. リスクベースプロセスアプローチ
4. 品質マネジメントシステム
820.20 経営者の責任 5. 経営者の責任
820.22 品質監査 8.2.4 内部監査
820.25 要員 6. 資源の運用管理
820.40 文書管理 4.2 文書化に関する要求事項
820.90 不適合製品 8.3 不適合製品の管理
820.100 是正処置及び予防処置 8.5 改善
820.250 統計的手法 8 測定、分析及び改善
B.製品実現フレームワーク
820.30 設計管理 7.3 設計・開発
820.50 購買管理 7.4 購買
820.60 識別 7.5.8 識別
820.65 トレーサビリティー 7.5.9 トレーサビリティー
820.70 生産及びプロセスの管理 7.5 製造及びサービスの提供
820.72 検査、測定及び検査装置 7.6 監視機器及び測定機器の管理
820.75 プロセスバリデーション 7.5.6 プロセスのバリデーション
820.80 受入、工程内及び完成機器のアクセプタンス
7.4.3 購買製品の検証
820.86 アクセプタンス状態 7.5.8 識別
820.120 機器のラベリング
820.130 機器の包装
820.140 取扱い 7.5.11 製品の保存
820.150 保管 7.5.11 製品の保存
820.160 流通 7.5.11 製品の保存
820.170 据付け 7.5.3 据付け活動
820.180 (記録) 4.2.5 記録の管理
820.181 DMR 4.2.3 医療機器ファイル
820.184 DHR 7.1 製品実現の計画
820.186 QSR 4.2.4 文書管理
820.198 苦情ファイル 8.2.2 苦情ファイル
820.200 附帯サービス 7.5.4 附帯サービス活動
FDA MDR&CAR HBコース Rev.10
リモート参加も可能なハイブリッド形式
〜 常に査察で確認される行政報告プロセス 〜
21CFR : Part 820, 803, 806
このコースの売り上げ金の一部は人道支援活動に寄付されます
☆ 開催概要 ☆
新しいQSRに変わるQMSRでISO13485にないプロセスを学び早期にプロセスを構築する必要があります。FDA査察レポートの解析からワースト4位(全体の約10%)に急浮上しているMDR(有害事象報告)、日本でも Warning Letter (警告書)を受けているほとんどの企業がMDRの指摘を受けています。昨今のデータ偽装事件を受け、FDAはこれらにフォーカスし厳しく査察を行っています。( 2021年レポート)。これはMDR報告の判断に迷ったら報告するという米国の文化とは異なり、日本のメーカーは情報を積極的に発信せず隠すという古い会社の文化が根強くあることが要因の一つですが、そもそも規制を理解せず正しい判断を行っていないことが大きな原因です。医療機器を作っているメーカーは、患者さんの命を守るという社会的な責任があります。会社を守っているようでも、結果的に会社が倒産に追い込まれることもある非常にリスクの高いプロセスであることを理解すべきです。それには規制の正しい理解、規制を守る仕組みと組織が必要です。このセミナーでは、CAPAサブシステム(苦情、不適合、CAPA)の各プロセスとリンクし、日本のメーカーの Warning Letter (警告書)を引き合いに正しいMDRプロセスと関連するCAR(リコール)プロセスを構築することを理解します。
このコースはオンサイトでも実施しています。お問い合わせはこちらから。
日 時:2023年3月6日(月曜)10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
リモートはZOOMから
対 象:既に医療機器・IVDを米国に輸出している企業
これから医療機器・IVDを米国輸出する準備を行っている企業
既にFDA査察を終え対応に困っている企業
受講料: ¥55,000-( 消費税込み、テキスト代含む、ご来場の方には昼食代含まれます )
リモートの方には事前にテキストを郵送させて頂きます
割 引: ¥44,000-( 消費税込み、テキスト代含む、ご来場の方には昼食代含まれます )
定 員: 20名( 会場 )リモート(30名)
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
特 割:弊社プロジェクトを実施中、又は継続研修をご希望の場合、御負担軽減のためさらに
割引をさせて頂きますのでお問い合わせください。この場合、正規料金でお申込の方
が優先され、出席できない場合がございますので予めご了承ください。
再受講:再受講をご希望の方はさらに割引が適用され、空席がある場合ご出席頂けます。
併せて過去ご出席なさった日をお知らせ下さい。
お申込:セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
リモートをご希望の方はその旨ご記入下さい。
開催要件:リモート参加の場合、リモート環境に確認とNDAへのサインをお願いしています。
パンデミックによる行政指導がある場合、ご来場の方もリモートに切り替えて頂き、
開催となります。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.規制を理解する
過去事例と統計からみる規制違反
理解すべき規制・規格・ガイダンス
eMDR規制
苦情組織と力量
規制が求めるあるべき組織と力量
プロセス構成
2.有害事象報告( MDR )Part 803
MDR規制の基本要件
FDA Warning letter から
規制の目的、重要性
MDRとして報告すべき事象とは
2.1 定義を理解する
重篤な障害
機能不全
報告すべき Malfunction
合理的に推測する
認識する
2.2 4.5日、30日報告
報告形態と時間軸
5日報告
ディシジョンツリー
2.3 FDAへの報告
報告フロー
報告様式 3500A
報告不要の場合
報告書の種類
報告免除
Warning letter 警告書
2.4 MDRプロセス
MDR報告フロー
CA,PAプロセス
3.回収報告(CAR)Part 806
規制の理解
報告免除
定義
リコールフロー
任意と強制リコール
定期メンテナンスとリコール
3.1 リコール判断基準
リコール判断基準例
ラベリングの定義
ラベリング違反と法的根拠
報告すべきラベリング違反例
3.2 CAR ディシジョンツリー
リコールディシジョンツリー
3.3 報告書と処理フロー
リコール判断後フロー
リコールクラス
有効性の確認
CAPAとの関係
4.警告書から理解すべき注意点とプロセスへの展開
Warning Letter ( 警告書 )の事例から理解する
警告書から理解すべき注意点とプロセスへの展開
プロセス構築後のレトロスペクティブな是正とは
*コース内容が一部変更される場合がございますが大きな変更はございません。

〜 FDA査察指摘ワースト キープロセス 〜
NEW QMSR 対応 21CFR Part 820.100、ISO13485 8.5
会場、リモート対応のハイブリッドコース!
このコースの売上金の一部は人道支援に寄付されます
* コース概要 *
CAPAプロセスを確認するだけで、その会社の品質システムや品質に対する理解度がよくわかります。CAPAプロセスに問題があれば、品質マネジメントシステムの適切性、妥当性はなく、特にマネジメントレビューがまともに運用されているとは到底思われません。品質マネジメントシステムの原則はプロセスアプローチ、PDCAをまわす為、マネジメントレビューのインプットでプロセスを監視し、改善ツールであるCAPAでプロセスを改善します。当たり前のことがシステムとして運用されていないことを理解している規制当局は、CAPAプロセスにフォーカスして確認しようとします。QMSが機能していないことは昨今の品質に関わる事件から明らかで、医療機器・IVDの場合、命に関わる大問題です。本コースでは今までの内容を大きく変更し、プロセスアプローチの手法に重点を置き、事例を元にCAPAの本質を理解して頂きます。 このコースは受講者と同じ目線の御社事例を用いることでより理解が高まるオンサイトセミナーが可能です。お問合せはこちらから。
日 時:2023年1月30日(月曜)9:30 〜 16:30
会 場:全国町村会館( 東京・永田町 ) ( 永田町駅から徒歩1分 )
リモート参加はZOOMで行います
講 師:クオリス・イノーバ株式会社代表取締役社長 木村 浩実
対 象:医療機器、IVD、医薬品等に限定せず、CAPAプロセスを学びたい企業様
受講料: ¥55.000-( 消費税込、テキスト、ご来場の方には昼食を提供 )
リモートでご参加なさる方には、事前にテキスト等を郵送致します
割 引: ¥44.000-( 消費税込、テキスト、ご来場の方には昼食を提供 )
1社2名様以上ご参加の場合
特 割: 弊社プロジェクト実施中、継続教育等の場合ご相談下さい。
定 員: クラス 定員 20名
リモート定員 30名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
開催要件:
1.リモート参加要件
会社からの場合、個室(会議室等)から会社PCで御参加となります。
ご自宅からの場合、会社PCでのご参加となります。
その他チェックリストを事前にお渡しし環境をご確認頂きます。
2.コロナウィルス対応
弊社のウィルス対策を順守して頂きます。
当日発熱がある場合、参加をお控え頂きます。
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.なぜFDAはCAPAに注目するのか
警告書の分析
2.なぜ品質問題は再発するのか
3.規制・規格を正しく理解する
QMSR/ISO
4.プロセスアプローチの基本
プロセス関連図
タートルチャート
5.TQMの仕組みが不可欠
マネジメントレビュー
PDCAをまわす
プロセス改善ツール
6.用語の正しい理解
修正、是正、予防の相違
逸脱、不適合、逸脱許可と特別採用の相違
修正、手直し、修理の相違
7.CAPAプロセスの IN・OUT
CAPAタートルチャート
不適合処理プロセス
苦情処理プロセス
CAPAプロセス
MDRプロセス
リコールプロセス
エスカレーションの仕組み
8.直接原因と根本原因を理解する
直接原因と根本原因の相違
根本原因の改善がプロセスアプローチの本質
9.予防プロセス
品質管理の原則、予防活動の理解
10.エスカレーション
CAPAエスカレーション
CAPAを上手に運用するための仕組み
11.CAPA7ステップ
Step 1 問題を検出し明確にする
Step 2 問題の原因を詳しく調査する
Step 3 根本原因の特定
Step 4 処置の特定と検証または妥当性確認
Step 5 予防または是正する為の処置
Step 6 処置の結果を文書化
Step 7 有効性の確認
12.SCARの実施
13.設計CAPA
14.CAPAマネジメント
マネジメントレビューの運用方法
15.品質問題の事例に学ぶ
Warning Letter Case Study
実際の事例
* 内容(項目)が変更される場合があります
 2021_4_CAPA course
2021_4_CAPA course
FDA QSR 内部監査員HBコース Rev.16
〜 QSR 対応内部監査員養成のための社内力量認定2日間教育コース 〜
* FDA QSIT をベースにプロセスアプローチで実施することを理解します *
このコースの売り上げ金の一部は人道支援活動に寄付されます
☆ 開催概要 ☆
最近の FDA 査察の資料によると、内部監査プロセスの指摘が多くなりつつある傾向で、FDA は内部監査の不備による指摘が多いと理解しています。不適合を出さないようプロセスに潜在する問題を未然に抽出し、マネジメントレビューでトップの指示のもとプロセス改善を常に行う。これがTQMの有るべき姿であり規制当局が求めている姿です。一方、現実はプロセスを監視し傾向管理を行っておらず、よく訓練されていないにわか監査員が規制・規格で要求されているからといって片手間に監査を行っているだけ。これは医療機器の場合致命的で、リコールで何十億の損失を出すだけではなく、人の命に関わる重大な問題に発展し会社の存続まで危ぶまれます。このコースでは、ISO内部監査員の資格を持つ方々がQSRの内部監査が行えるよう社内資格を付与するための社内資格認定コースとして位置づけられます。一般に行われているISOの内部監査手法とは異なる、付加価値を生む予防対策としての監査手法とノウハウを学びます。( このコースを受講した後、オンサイトでクオリス・イノーバによる内部監査のOJTを行うとより効果的です )
日 時:2022年12月5日(月曜)、6日(火曜) 9:30 〜 16:30
会 場: 全国町村会館(東京・永田町) 永田町駅から徒歩1分( 3番出口 )
リモートでご参加はZOOMとなります
人数が少ない場合でも開催しますが、リモートのみになる可能性もございますので
予めご了承いただきますようお願いいたします。
対 象:すでに弊社主催下記コースをオープン、オンサイト問わず受講された方限定です。
* FDA GMP QSR 2日間コース
* FDA 設計管理2日間コース
このコースはQSRの基礎知識が無いと受講できず、弊社のコースを受講されていない場合
異なる解釈により内容をよく理解出来ません。 何卒ご理解をお願い申し上げます。
受講料: ¥99,000( 税込、テキスト・昼食含む )
割 引: ¥88,000( 税込、テキスト・昼食含む )
1社2名様以上受講される場合
定 員: 20名限定 満席になり次第締切ます
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
過去に受講された弊社QSR、設計管理各コースの受講日を記載して下さい。
オンサイトで受講された場合でも構いません。
折り返し、会場、お振込先情報をメールでご連絡致します。
セミナーに参加するための宿題を追って発送し致します。
2名様以上ご参加の場合、お名前とローマ字読み、
メールアドレスを連絡欄にご記入下さい。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.内部監査の目的を理解する
規制・規格が求めていること
患者の命を優先する監査とは
企業の付加価値を高める為には
2.内部監査員に求められる力量
内部監査員とリーダーに求められる力量
内部監査員訓練
マネジメントチームのカウンセラー
3.プロセスアプローチ
CAPAの理解とCAPAアプローチ
プロセスのR&R( 役割と責任 )を明確にする
プロセスを常に測定し改善する
内部監査はプロセスに注目
4.監査判定基準
チェックリストと数値化
プロセス毎に数値化
FDA査察と内部監査判断基準の相違を知る
5.監査を計画する
マネジメントレビューと品質会議からの指示
リスクベース&プロセスアプローチによる監査計画
プロセス管理パラメートと傾向分析結果
是正監査と予防監査の相違
監査は短期集中型で実施
監査員レビュー会のノウハウ
監査報告書記載のポイント
6.内部監査レポート
トップマネジメントに対する報告
プロセスの不足、改善点を報告
規制に対するコンプライアンス状況の報告
トップマネジメントのアクション
7.監査の目のつけどころ( QSIT )
QSITに基づく監査手法と勘所
プロセスアプローチの手法を全員が理解
1.経営者による管理
2.設計管理
3.CAPA
MDR報告
CAR
MDトラッキング
4.P&PC
5.滅菌工程
+6.各プロセス
*内容は変更される場合がございます*
2017_12_Internal auditor
QMS 購買プロセスHBコース
〜 FDA 査察で指摘 Worst 4 購買プロセスの本質とは 〜
21CFR Part 820・ISO13485・ISO9001
このコースの売り上げ金の一部は人道支援活動に寄付されます
会場参加、リモート、ハイブリッド形式で開催されます
弊社コロナウィルス対策はこちらからご確認下さい
☆ 開催概要 ☆
購買プロセスは言われた通りにモノを買っていればよいというものではなく、サプライチェーン全体を俯瞰し、製品企画時の上流から積極的にコンカレントで参画、最も効率よいQCDを提案することこそ会社に貢献するプロセスとして価値があります。その為には、戦略的な組織体制とフレームを構築する必要があり、開発購買や情報購買と言ったフレームワークは早期の製品ローンチを実現します。また、アッセンブルラーが多くなった昨今、行政当局も購買プロセスに多くの査察時間を費やすようになりました。自社と同じシステムで管理する必要性が高まり、フレームと管理能力が要求されます。FDA QSRのみならず、ISOの基本9001の要求も加味しながら理解を深めます。
オンサイトでも可能です。お問い合わせはこちらから。
日 時:2022年11月28日(月曜) 9:30 〜 16:30
会 場:全国町村会館(東京・永田町) 永田町駅から徒歩1分( 3番出口 )
リモートでご参加なさる方は ZOOM でのご参加となります
講 師:クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
ODM、OEMとして設計・製造を受託している企業
受講料: ¥55.000-( 消費税込、テキスト、ご来場の方には昼食を提供 )
リモートでご参加なさる方には、事前にテキスト等を郵送致します
割 引: ¥44.000-( 消費税込、テキスト、ご来場の方には昼食を提供 )
1社2名様以上ご参加の場合
特 割: 弊社プロジェクト実施中、継続教育等の場合ご相談下さい。
定 員: クラス 定員 20名
リモート定員 30名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
開催要件:
1.リモート参加要件
会社からの場合、個室(会議室等)から会社PCで御参加となります。
ご自宅からの場合、会社PCでのご参加となります。
その他チェックリストを事前にお渡しし環境をご確認頂きます。
2.コロナウィルス対応
弊社のウィルス対策を順守して頂きます。
当日発熱がある場合、参加を御控え頂きます。
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
A.FDA規制、査察の最新情報と傾向分析(QMSRの動向)
1.適用規制・規格
FDA QSR,ISO13485,9001の要求とは
プロセスアプローチの購買プロセスとは
購買プロセスの位置づけ
2.購買管理の目的
規格が示す購買管理の目的
製品、サービス購買とは
4つの購買範囲とは
3.用語の定義
製品購買、サービス購買とは
プロバイダー、サプライヤー、コントラクターとは
4.購買管理
GHTF( IMDRF )ガイダンスのフロー
5.購買方針
購買方針がなければ購買プロセスは成り立たない
6.戦略・開発購買
サプライチェーン全体を俯瞰した戦略
開発スピードを上げ原価削減、購買活動のスピードを上げある
ベストプラクティスの収集と技術の取り込み
7.購買計画
製品企画段階から参画
供給する製品、サービスの特定
技術及びプロセス情報の特定
供給者候補の特定
リスク、管理方法の特定
8.供給者候補選定
供給者ビジネス、運用能力評価
供給者候補選定
9.供給者選定
評価選定基準の策定
供給者候補の能力評価
供給者の承認
10.管理方法の取り決め
購買情報とは
管理方法
11.変更時対応
変更管理規定
4Mと変化点管理
12.納期、測定、監視
製品、サービスの検証
不適合の手順
供給者の監視
製品の受領、出荷場、冷蔵室、NCエリア
カーゴ内温度バリデーション
13.フィードバック、コミュニケーション
CR
SCAR
供給者評価シート
14.サプライヤー監査
リスクベース監査
5つの監査、評価エリア
情報購買、戦略上重要な監査チーム
( 内容が変更になる場合もございますが大きな変更はございません。)
2021_11_PC
FDA 設計管理2日間HBコース Rev.27
〜 リスクをヘッジしコンプライアンスを堅持するフレームワーク 〜
NEW QMSR 対応 21CFR Part 820.30 / 75 ISO13485
QSR内部監査員コースご出席要件コースです
( オンサイト及び過去弊社コースを受講された方は要件を満たします )
会場参加、リモート、ハイブリッド形式で開催します
☆ 開催概要 ☆
ほとんどの日本企業に於ける医療機器開発・設計管理の仕組みは世界標準にほど遠く、これはグローバルに進出しようとする企業のみならず、患者の命を預かる全ての医療機器メーカーにとって大きなリスクを伴います。米国FDAは自らの査察で、医療機器品質不良の根本原因は上流側の設計管理にあると査察の結果から統計的に見抜いており、それはクオリス・イノーバの10年以上にわたるGAP監査でも明らかです。そこで本コースでは、グローバルな開発、設計管理を米国FDA「設計管理ガイダンス」及び欧米で取り入れられている開発フレームワークを元に体系的に解説、また、品質不良を発生させない仕組み(ロバスト設計)をタグチメソッドと設計CAPAを用いて解説します。 2日目はプロセスバリデーションの本質、工程内試験に頼らない均一で高品質な製品を一貫して製造する為、パラメータによる製造方法を実現する工程設計について解説します。 ”規制” だからではなく、設計品質を高めることにより患者さんの命を守り、企業にとっても大きな潜在的利益を得られるという大原則は、エンジニアの方々から多くの共感と支持を頂いてきたクオリス・イノーバの最も人気のある価値あるコースです。 このコースはオンサイトでも実施しております。お問い合わせはこちらから。
日 時: 2022年11月7日(月曜)、8日(火曜) 9:30〜16:30
締 切: 2022年11月1日(火曜)
会 場: 全国町村会館 大ホール(東京・永田町) 永田町駅から徒歩1分( 3番出口 )
リモートでご参加はZOOMとなります
講 師: クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象: 医療機器・IVD市場にこれから参入しようとしている企業
医療機器・IVDを海外に輸出しようとしている企業
FDA査察準備、MDSAPを受審しようと準備している企業
既にFDA査察を終え、指摘対応に苦慮している企業
製品実現に関わる各組織( マーケティング、開発、生産技術、品証など )
受講料: ¥99,000( 税込、テキスト・昼食含む )
割 引: ¥88,000( 税込、テキスト・昼食含む )
1社2名様以上受講される場合
特 割: 再受講の方、プロジェクト終了企業様で継続研修としてご検討の方ご相談下さい。
定 員: クラス 定員 20名
リモート定員 30名
出席者全員に教育記録として利用できる修了書をお渡しします
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡いたします。
2名様以上ご参加の場合、お名前とローマ字読み、メールアドレスを連絡欄にご記入下さい。
持参品: 弊社QSR翻訳本第5版をご持参下さい。お持ちでない方はこちらからお求め頂けます。
ご注意: 本コースは、品質マネジメントシステムの一部を解説するため、システム全体の知識がないと理解できません。本コース受講前に5月開催QMSR2日間コースの受講をお勧めします。( QMSR2日間コースで訳本を配布しています )
開催要件:
1.コロナウイルス対応
行政の指示による規制の対象となった場合、リモートのみの開催となります。
弊社のウィルス対策を順守して戴きます。
2.リモートご参加要件
ZOOM以外での対応はしておりません
会社からご参加の場合、会議室から会社PCでのご参加となります。
自宅からご参加の場合、会社PCでのご参加となります。
その他、チェックリストでご確認頂く為、事前にご確認をお願いしています。
メールでお問い合わせ
電話でお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1日目(セミナー内容)
A.コンプライアンスとインテグリティー
設計不良の原因は何か
設計CAPAとは
ケーススタディ
不正を排除する仕組み
規制・規格に満足するればOKか?
人の命
行政によるミッションの相違
B.医療機器規制を理解する
新しい規則QMSRと今後の対応値
適用規制、規格、ガイダンス
1.Subpart C 設計管理 820.30 ISO13485 - 7.3
設計管理の目的とその重要性
プロセスアプローチの枠組と製品実現フレームワークの重要性
ライフサイクルマネジメント(LCM)と Waterfall Model
関連する ISO13485 の項目(プロセス)
Form 483 の FY 統計的解析から理解すること
2.820.30 (a) 総則 ISO13485 - 7.3.1
プロセスアプローチ ISO13485 4.1 の理解と
経営者の責任(リソースの提供とKPI)
製品実現プロセス
タートルチャート
リソースとしての力量(教育と訓練) 820.25
文書体系から俯瞰する設計管理プロセス
3.820.30 (b) 設計及び開発計画 ISO13485 - 7.3.2
医療機器開発ライフサイクルマネジメント(LCM)
設計品質確立の重要な要素; コンカレントエンジニアリング(CE)
PM、PLの役割と責任
ライフサイクルリスクマネジメントの理解(LCRM) ISO14971
タグチメソッド開発フレームワークによるフロントローディング手法
マーケティング戦略の重要性
開発計画書
4.820.30 (c) 設計インプット ISO13485 - 7.3.3
コミュニケーションエラー
曖昧なニーズの明確化
要求仕様から完成品までの一貫性
トレーサビリティーマトリックス(TMX)
ラベリング、包装設計のインプット
5.820.30 (d) 設計アウトプット ISO13485 - 7.3.4
タートルチャートのアウトプット
設計アウトプットのリスト
E−BOMの役割
製造仕様書とは
6.820.30 (e) 設計レビュー ISO13485 - 7.3.5
問題の早期発見、解決が目的でセレモニーで終わらせない仕組み
設計品質確立の重要な要素
設計レビューとテクニカルレビュー
レビューワーと責任と権限
7.820.30 (f) 設計検証 ISO13485 - 7.3.6
インプットからアウトプットまでのトレーサビリティー
製品の安全性、性能を証明するエビデンスの作り方
設計検証計画に必要な要素
設計検証レポートはデータの信頼性を確保
8.820.30 (g) 設計バリデーション ISO13485 - 7.3.7
設計検証と設計バリデーションの相違
設計バリデーションの手法
2つのバリデーションの相違と記録
ソフトウェアのバリデーションとVモデル
3種類のバリデーションの相違
9.820.30 (h) 設計移管 ISO13485 - 7.3.8 → 第2日目(工程設計)
10.820.30 (i ) 設計変更 ISO13485 - 7.3.9
変更管理プロセスに伴う設計変更
ライフサイクルリスクマネジメント
タートルチャート
設計変更フロー
11.820.30 (i ) 設計変更 ISO13485 - 7.3.10 設計・開発ファイル
DHF / DMR / DHR の定義
識別、ファイルの基本
文書体系と設計標準
2日目(セミナー内容)
設計移管と工程設計
820.30 (h)、13485 - 7.3.8、7.5.6
1.820.30 (h) 設計移管 ISO13485 - 7.3.8
ライフサイクルリスクマネジメントにおける設計移管(工程設計)
均一な製品を一貫して生産する手法
プロセスバリデーションの目的
2.なぜ工程バリデーションを実施するのか 820.75, ISO13485 - 7.5.6
プロセスバリデーションの目的とその本質
プロセスバリデーションの効果
工程管理、監視の目的と本質
医療機器規制・規格の理解
3.バリデーションすべき工程とは
バリデーションすべき工程の識別
検証工程とバリデーション工程の相違
設備バリデーションと工程バリデーション
バリデーション工程例
4.バリデーションの種類
バリデーションの種類
設備バリデーションと劣化パラメータ 820.70(g), 72(a)
コンピューターシステムバリデーション(CSV) 820.70(i), ISO13485 - 7.5.6 (g)
5.バリデーションステップ 820.75, ISO13485 - 7.5.6
VMP:バリデーションマスタープラン
DQ: 設計時適格性評価
IQ: 据付時適格性評価
OQ: 稼働時適格性評価
PQ: 製造時適格性評価
PPQ: プロセス街道性評価
バリデーション文書化
プロセスバリデーションの明確化
自動化工程の明確化
CSVカテゴリーの明確化
ソフトウェアのバリデーションの基本
リスクベースCSV
6.工程設計とプロセスバリデーション 820.30 (h)、 ISO13485 - 7.3.8
ライフサイクルマネジメントに於ける工程設計
工程設計の進め方
工程設計とプロセスリスク分析( PFMEA )
環境管理 820.70(c)、 ISO13485 - 6.4
汚染管理 820.70(e)、 ISO13485 - 6.4
副資材 820.70(h)、 ISO13485 - 7.5.2
工程設計PTMX
QC工程表
7.回顧的バリデーション
8.バリデーションのアウトプット
QC工程表と統計的手法(工程能力)
リバリデーション設定
設備インスペクションとメンテナンス
( 内容が変更される場合がございます )
2022_06 設計管理2日間コース
付帯サービス リモートコース
〜 医療機器グローバルサービス設計とデータ運用 〜
21CFR Part 820.200 / ISO13485 7.5.4
このコースの売上金の一部は人道活動支援に寄付されます
☆ 開催概要 ☆
ほとんどの日本企業に於ける医療機器付帯サービス活動は製品としてビジネスが成立していません。また、プロセスとしてコンカレント設計がない企業ではサービス活動は製品設計のインプットになく、付帯サービスで無駄なコストや製品価値の低下を招き、グローバルにもサービス品質で競争に負けています。さらに、サービスデータは開発の上流にフィードバックすべきデータの宝庫で、改善の積み重ねが品質を高め、結果として患者さんの命を守ります。これらは規制、規格でも求められています。
オンサイトでも可能です。お問い合わせはこちらから。
日 時: 2022年10月24日(月曜)13:30 〜 16:30 午後半日コースです
会 場: リモートのみ ZOOMでのご参加となります
講 師: クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象: 医療機器・IVD市場にこれから参入しようとしている企業
医療機器・IVDを海外に輸出しようとしている企業
FDA査察準備、MDAPを受審しようと準備している企業
既にFDA査察を終え、指摘対応に苦慮している企業
受講料: ¥38,500( 消費税込、テキスト含む )
割 引: ¥33,000( 消費税込 キスト含む )
1社2名様以上受講される場合
特 割: 企業で実施しているQMS構築プロジェクトや弊社が実施する継続研修などで受講
なさる場合、特別割引きを適用させて頂きますのでメールでご相談下さい。
定 員: 30名
出席者全員に教育記録として利用できる修了書をお渡しします
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡いたします。
2名様以上ご参加の場合、お名前とローマ字読み、メールアドレスを連絡欄にご記入下さい。
参考品: 弊社QSR翻訳本第5版をお勧めします。お持ちでない方はこちらからお求め頂けます。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.FDA規制、査察の最新情報と傾向分析
規制当局の警告書分析と傾向
2.医療機器規制・規格の理解
適用規制・規格
品質計画( 付帯サービスプロセス )
プロセスアプローチKPI
タートルチャート
3.付帯サービスの目的の理解
付帯サービスの目的
QSRとISO、省令から理解する
言葉の定義
4.サービス設計
ライフサイクルサービス活動の設計
820.30 の適用
コンカレント設計とは
設計アウトプット(サービス向け)
5.ライフサイクルマネジメント
PM1:製品開発の上流から参画しサービス設計実施
ライフサイクルマネジメントサービス活動
設計へのインプット
サービス要求仕様書
サービス活動のリモート化
M2 :サービス診断ツールの検証
サービス効率化に欠かせない診断ツール
診断ツールの検証
M3 :サービスマニュアル検証とバリデーション
マニュアルの検証とバリデーション
サービストレーニング
インスタレーション規制の理解
M4 :パーツハンドリング
サービスパーツの保管
S−BOM
劣化製品の取扱
6.有害事象報告(MDR)
サービス報告と苦情
MDRとは
MDR提出判断とフロー
7.統計的傾向監視
統計的分析
CAPA
上流へのフィードバック
FDA QMSR/QSR/13485 2日間リモートコース
〜 医療機器・IVDに求められるQMSの本質を学ぶ 〜
21CFR Part 820
このコースはリモートのみ開催です。会場出席をご希望の方は
9月29日、30日開催HBコースへご参加下さい。
2022年3月に公開された QSR に変わる新たな規制 QMSR について、
ISO13485 との相違はもとより、原点に立ち返る QMS の本質について学ぶ新たなコースです。
本コースは、12月に実施する監査員コースの受講要件のための特別コースで
9月受講できなかった方もリモートでご参加頂けます!
開催内容は、9月開催と同様ですのでこちらをご参照下さい。
☆ 開催概要 ☆
QSR翻訳本(第5版)+QMSR翻訳本(第1版)付き
日 時: 2022年10月20日(木曜)、21日(金曜) 9:30〜16:30
会 場: リモートのみ ZOOMでのご参加となります。
講 師: クオリス・イノーバ株式会社
取締役社長 木村 浩実
対 象: 医療機器・IVD市場にこれから参入しようと考えている企業
米国に輸出を計画している企業
米国FDA 査察準備を考えている企業
ODM、OEM として医療機器を受託しようとしている企業
受講料: ¥99,000−(税込)( 2日分、テキスト、対訳本2冊 )
事前にテキスト、対訳本を郵送いたします。
割 引: ¥88,000−(税込)
1社2名以上ご参加の場合
特 割: 企業で行うQMS構築プロジェクトなどや企業で弊社が実施した継続研修などで
ご利用の場合、特別割引を適用させて頂きますのでメールでご相談下さい。
満席となった場合、正規料金をお支払いの方が優先となりご希望に添えない場合
がございますので予めご了承ください。
定 員: リモート定員 30名様
( 出席者全員に教育記録として利用できる修了書をお渡しします )
開催要件:
リモート参加要件
会社からの場合、個室(会議室等)から会社PCで御参加となります。
ご自宅からの場合、会社PCでのご参加となります。
その他チェックリストを事前にお渡しし環境をご確認頂きます。
お申込: セミナー申込みページからお申し込み下さい。
折り返し、会場、お振り込み先情報などをメールでご案内致します。
2名以上で参加なさる方ははその旨ご記入下さい。
割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
FDA QMSR/QSR/13485 2日間HBコース
〜 医療機器・IVDに求められるQMSの本質を学ぶ 〜
21CFR Part 820
このコースは、オンサイト、リモートのハイブリッド開催です。
2022年3月に公開された QSR に変わる新たな規制 QMSR について、
ISO13485 との相違はもとより、原点に立ち返る QMS の本質について学ぶ新たなコースです。
年末に実施する監査員コースの受講要件となります。
☆ 開催概要 ☆
QSR翻訳本(第5版)+QMSR翻訳本(第1版)付き
米国が最も大きなマーケットであることは医療機器でも例外では無く、米国での成功がグローバルを制すると言っても過言ではありません。しかし、米国で製品を上市するには医療機器規制 QMSR に基づく QMS の構築は必須となります。2023年から新たに適用となるこの規制は、FDA QSR 規制を積極的に取り入れてきた ISO13485(2016) にほぼ準拠となりますが、ガイダンスなどは今まで通り適用となります。今後は QSR の欠点でもあった QMS の本質、リスクベースプロセスアプローチのフレームワークが必須となります。弊社の経験上 ISO1345 取得済みの企業であっても ISO に準拠しているとはとても言いがたい状況で QMSR には準拠できませんのでご注意下さい。今まで弊社は一貫して規制の本質を解き、事業活動とのインテグレーションを取ることにより、結果の出せるマネジメントシステムを提唱し、多くの企業の皆様の共感を集め支持されて参りました。米国に進出する企業のみならず、医療機器・IVDを開発しようとしている企業の皆様のマインドが大きく変わる価値あるクオリス・イノーバのメインコースです。
新規制が適用となるまで QSR が適用となる為、本コースでは現行 QSR, QSMR, ISO13485 について、リスクベースプロセスアプローチ、製品実現フレームワークといったシステムの根幹となるフレームワークを中心に概要を説明します。
オンサイト(一定人数が集まった会社でのセミナー)も可能です。お問合せはこちらから。
日 時: 2022年9月26日(月曜)、27日(火曜) 9:30〜16:30
会 場: 全国町村会館(東京・永田町)(永田町駅から徒歩1分)
リモートで参加なさる場合、ZOOMでの御参加となります。
講 師: クオリス・イノーバ株式会社
取締役社長 木村 浩実
対 象: 医療機器・IVD市場にこれから参入しようと考えている企業
米国に輸出を計画している企業
米国FDA 査察準備を考えている企業
ODM、OEM として医療機器を受託しようとしている企業
受講料: ¥99,000−(税込)( 2日分、テキスト、対訳本2冊、昼食代含 )
リモートで参加なさる方には事前にテキスト、対訳本を郵送いたします。
割 引: ¥88,000−(税込)
1社2名以上ご参加の場合
特 割: 企業で行うQMS構築プロジェクトなどや企業で弊社が実施した継続研修などで
ご利用の場合、特別割引を適用させて頂きますのでメールでご相談下さい。
満席となった場合、正規料金をお支払いの方が優先となりご希望に添えない場合
がございますので予めご了承ください。
定 員: クラス 定員 20名様 9月20日締切
リモート定員 30名様
( 出席者全員に教育記録として利用できる修了書をお渡しします )
開催要件:
1.リモート参加要件
会社からの場合、個室(会議室等)から会社PCで御参加となります。
ご自宅からの場合、会社PCでのご参加となります。
その他チェックリストを事前にお渡しし環境をご確認頂きます。
2.コロナウィルス対応
弊社のウィルス対策を順守して頂きます。
当日発熱がある場合、参加を御控え頂きます。
お申込: セミナー申込みページからお申し込み下さい。
折り返し、会場、お振り込み先情報などをメールでご案内致します。
2名以上で参加なさる方ははその旨ご記入下さい。
割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.コンプライアンスとインテグリティー
患者の命と会社の危機管理
データインテグリティー
2.医療機器規制の理解
行政によるミッションの相違
米国規制法
医療機器規制の理解
3.QSR から QMSR へ移行する経緯
4.QSR と QMSR 、ISO13485( 2016) との相違
5.QMSR 移行後変わるものと変わらないもの
6.なぜ ISO 認証では FDA 査察にパスしないのか
7.QSIT アプローチとは
8.QMSR
Subpart A 総則
820.1 適用範囲
820.3 定義
規制を正しく理解する要素:定義
820.7 参照による引用
規制に記載していない ISO の引用について
820.10 品質マネジメントシステムに関する要求事項
ISO13485(2016) 適用の要件
820.15 コンセプトの明確化
Subpart B 附則
820.35 記録の管理
820.45 機器のラベリング及び包装管理
9.QMS を構成するフレームワーク
QSR ISO13485 (2016)
A. リスクベースプロセスアプローチ
4. 品質マネジメントシステム
820.20 経営者の責任 5. 経営者の責任
820.22 品質監査 8.2.4 内部監査
820.25 要員 6. 資源の運用管理
820.40 文書管理 4.2 文書化に関する要求事項
820.90 不適合製品 8.3 不適合製品の管理
820.100 是正処置及び予防処置 8.5 改善
820.250 統計的手法 8 測定、分析及び改善
B.製品実現フレームワーク
820.30 設計管理 7.3 設計・開発
820.50 購買管理 7.4 購買
820.60 識別 7.5.8 識別
820.65 トレーサビリティー 7.5.9 トレーサビリティー
820.70 生産及びプロセスの管理 7.5 製造及びサービスの提供
820.72 検査、測定及び検査装置 7.6 監視機器及び測定機器の管理
820.75 プロセスバリデーション 7.5.6 プロセスのバリデーション
820.80 受入、工程内及び完成機器のアクセプタンス
7.4.3 購買製品の検証
820.86 アクセプタンス状態 7.5.8 識別
820.120 機器のラベリング
820.130 機器の包装
820.140 取扱い 7.5.11 製品の保存
820.150 保管 7.5.11 製品の保存
820.160 流通 7.5.11 製品の保存
820.170 据付け 7.5.3 据付け活動
820.180 (記録) 4.2.5 記録の管理
820.181 DMR 4.2.3 医療機器ファイル
820.184 DHR 7.1 製品実現の計画
820.186 QSR 4.2.4 文書管理
820.198 苦情ファイル 8.2.2 苦情ファイル
820.200 附帯サービス 7.5.4 附帯サービス活動
2022-05 QSR seminar
FDA MDR&CAR HBコース Rev.9
リモート参加も可能なハイブリッド形式
〜 新QMSRを理解するプロセス第二弾 〜
21CFR : Part 820, 803, 806
このコースの売り上げ金の一部は人道支援活動に寄付されます
☆ 開催概要 ☆
新しいQSRに変わるQMSRでISO13485にないプロセスを学び早期にプロセスを構築する必要があります。FDA査察レポートの解析からワースト4位(全体の約10%)に急浮上しているMDR(有害事象報告)、日本でも Warning Letter (警告書)を受けているほとんどの企業がMDRの指摘を受けています。昨今のデータ偽装事件を受け、FDAはこれらにフォーカスし厳しく査察を行っています。( 2021年レポート)。これはMDR報告の判断に迷ったら報告するという米国の文化とは異なり、日本のメーカーは情報を積極的に発信せず隠すという古い会社の文化が根強くあることが要因の一つですが、そもそも規制を理解せず正しい判断を行っていないことが大きな原因です。医療機器を作っているメーカーは、患者さんの命を守るという社会的な責任があります。会社を守っているようでも、結果的に会社が倒産に追い込まれることもある非常にリスクの高いプロセスであることを理解すべきです。それには規制の正しい理解、規制を守る仕組みと組織が必要です。このセミナーでは、CAPAサブシステム(苦情、不適合、CAPA)の各プロセスとリンクし、日本のメーカーの Warning Letter (警告書)を引き合いに正しいMDRプロセスと関連するCAR(リコール)プロセスを構築することを理解します。今回は薬機法のGVP規制とのリンクも解説します。
このコースはオンサイトでも実施しています。お問い合わせはこちらから。
日 時:2022年9月5日(月曜)10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
リモートはZOOMから
対 象:既に医療機器・IVDを米国に輸出している企業
これから医療機器・IVDを米国輸出する準備を行っている企業
既にFDA査察を終え対応に困っている企業
受講料: ¥55,000-( 消費税込み、テキスト代含む、ご来場の方には昼食代含まれます )
割 引: ¥44,000-( 消費税込み、テキスト代含む、ご来場の方には昼食代含まれます )
定 員: 12名( 会場 )リモート(20名)
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
特 割:弊社プロジェクトを実施中、又は継続研修をご希望の場合、御負担軽減のためさらに
割引をさせて頂きますのでお問い合わせください。この場合、正規料金でお申込の方
が優先され、出席できない場合がございますので予めご了承ください。
再受講:再受講をご希望の方はさらに割引が適用され、空席がある場合ご出席頂けます。
併せて過去ご出席なさった日をお知らせ下さい。
お申込:セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
リモートをご希望の方はその旨ご記入下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.規制を理解する
過去事例と統計からみる規制違反
理解すべき規制・規格・ガイダンス
eMDR規制
苦情組織と力量
規制が求めるあるべき組織と力量
プロセス構成
2.有害事象報告( MDR )Part 803
MDR規制の基本要件
FDA Warning letter から
規制の目的、重要性
MDRとして報告すべき事象とは
2.1 定義を理解する
重篤な障害
機能不全
報告すべき Malfunction
合理的に推測する
認識する
2.2 4.5日、30日報告
報告形態と時間軸
5日報告
ディシジョンツリー
2.3 FDAへの報告
報告フロー
報告様式 3500A
報告不要の場合
報告書の種類
報告免除
Warning letter 警告書
2.4 MDRプロセス
MDR報告フロー
CA,PAプロセス
3.回収報告(CAR)Part 806
規制の理解
報告免除
定義
リコールフロー
任意と強制リコール
定期メンテナンスとリコール
3.1 リコール判断基準
リコール判断基準例
ラベリングの定義
ラベリング違反と法的根拠
報告すべきラベリング違反例
3.2 CAR ディシジョンツリー
リコールディシジョンツリー
3.3 報告書と処理フロー
リコール判断後フロー
リコールクラス
有効性の確認
CAPAとの関係
4.警告書から理解すべき注意点とプロセスへの展開
Warning Letter ( 警告書 )の事例から理解する
警告書から理解すべき注意点とプロセスへの展開
プロセス構築後のレトロスペクティブな是正とは
*コース内容が一部変更される場合がございますが大きな変更はございません。

2022_3_MDR/CAR
〜 QSRに変わる新しい規制の解説 〜
ご要望にお応えし再度開催します
☆ 開催概要 ☆
FDAが3月2日に実施したQSRに変わる新しい規制QMSRのカンファレンスの内容について概要を説明するショートセッションです。リモートのみでの開催となります。
1.QMSRのロードマップ
2.CFR Part 820 の扱い
3.QSRとQMSR、ISO13485の相違
4.今後のFDA査察
日 時: 7月21日(木曜) 15時〜16時
会 場: リモートのみ( ZOOM )
講 師: クオリス・イノーバ株式会社
代表取締役社長 木村 浩実
受講料: 無料
定 員: 50名様限定( 再受講も可能 )
お申込: 下記よりお申込ください。追ってZOOMのリンクをお知らせします。
ご注意: ZOOM以外では実施しておりません。
氏名表示し、カメラで参加者が確認できる必要があります。
カメラ映像は他の参加者には映らないモードで実施します。
書 籍: 事前に下記の飜訳本+ワンポイント解説があると理解が進みます
QMSR翻訳本第0版
ご購入はこちらから
メールでのお問い合わせ

QMSR ショートセッション
〜 QSRに変わる新しい規制の解説 〜
ご要望にお応えし再度開催します
☆ 開催概要 ☆
FDAが3月2日に実施したQSRに変わる新しい規制QMSRのカンファレンスの内容について概要を説明するショートセッションです。リモートのみでの開催となります。
1.QMSRのロードマップ
2.CFR Part 820 の扱い
3.QSRとQMSR、ISO13485の相違
4.今後のFDA査察
日 時: 6月21日(火曜) 15時〜16時
会 場: リモートのみ( ZOOM )
講 師: クオリス・イノーバ株式会社
代表取締役社長 木村 浩実
受講料: 無料
定 員: 80名様限定( 再受講も可能 )
お申込: 下記よりお申込ください。追ってZOOMのリンクをお知らせします。
ご注意: ZOOM以外では実施しておりません。
氏名表示し、カメラで参加者が確認できる必要があります。
カメラ映像は他の参加者には映らないモードで実施します。
書 籍: 事前に下記の飜訳本+ワンポイント解説があると理解が進みます
QMSR翻訳本第0版
ご購入はこちらから
メールでのお問い合わせ
〜 誰も教えないプロセスアプローチのツール 〜
NEW QMSR 対応 21CFR Part 820.100、ISO13485 8.5
会場、リモート対応のハイブリッドコース!
このコースの売上金の一部は人道支援に寄付されます
* コース概要 *
CAPAプロセスを確認するだけで、その会社の品質システムや品質に対する理解度がよくわかります。CAPAプロセスに問題があれば、品質マネジメントシステムの適切性、妥当性はなく、特にマネジメントレビューがまともに運用されているとは到底思われません。品質マネジメントシステムの原則はプロセスアプローチ、PDCAをまわす為、マネジメントレビューのインプットでプロセスを監視し、改善ツールであるCAPAでプロセスを改善します。当たり前のことがシステムとして運用されていないことを理解している規制当局は、CAPAプロセスにフォーカスして確認しようとします。QMSが機能していないことは昨今の品質に関わる事件から明らかで、医療機器・IVDの場合、命に関わる大問題です。本コースでは今までの内容を大きく変更し、プロセスアプローチの手法に重点を置き、事例を元にCAPAの本質を理解して頂きます。 このコースは受講者と同じ目線の御社事例を用いることでより理解が高まるオンサイトセミナーが可能です。お問合せはこちらから。
日 時:2022年7月4日(月曜)9:30 〜 16:30
会 場:全国町村会館( 東京・永田町 ) ( 永田町駅から徒歩1分 )
リモート参加はZOOMで行います
講 師:クオリス・イノーバ株式会社代表取締役社長 木村 浩実
対 象:医療機器、IVD、医薬品等に限定せず、CAPAプロセスを学びたい企業
受講料: ¥55,000 消費税込( テキスト代・昼食含む(ご来場の方のみ) )
割 引: ¥44,000 消費税込( テキスト代・昼食含む(ご来場の方のみ) )
1社2名様以上ご参加の場合
特 割:弊社プロジェクトを実施中の場合や、継続研修をご希望の場合、ご負担軽減のためさらに
割引をさせて頂きますのでお問い合わせください。 この場合、正規料金をお支払いのお
客様が優先され、ご出席できない場合がございますのでご了解ください。
再受講:空席がある場合、半額で再受講可能ですのでお問い合わせください。
併せて受講された日もご記入ください。
定 員: クラス :20名様
リモート:30名様 6月27日締め切り
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
締 切:6月27日締め切り
開催要件;コロナウィルスの対応について
1.換気は自動換気システムが作動し入口の扉を開けて開催します
2.入口にアルコールを設置、手指消毒と服、靴ウラの消毒をお願いします
3.テーブル、席はホテル側で消毒が行われます
4.スタッフ、出席者全員マスク着用でお願いいたします
お申込:セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールで連絡いたします。
複数名ご参加の場合、同様の情報をご記入下さい。 割引料金でご案内いたします。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.なぜFDAはCAPAに注目するのか
警告書の分析
2.なぜ品質問題は再発するのか
3.規制・規格を正しく理解する
QMSR/ISO
4.プロセスアプローチの基本
プロセス関連図
タートルチャート
5.TQMの仕組みが不可欠
マネジメントレビュー
PDCAをまわす
プロセス改善ツール
6.用語の正しい理解
修正、是正、予防の相違
逸脱、不適合、逸脱許可と特別採用の相違
修正、手直し、修理の相違
7.CAPAプロセスの IN・OUT
CAPAタートルチャート
不適合処理プロセス
苦情処理プロセス
CAPAプロセス
MDRプロセス
リコールプロセス
エスカレーションの仕組み
8.直接原因と根本原因を理解する
直接原因と根本原因の相違
根本原因の改善がプロセスアプローチの本質
9.予防プロセス
品質管理の原則、予防活動の理解
10.エスカレーション
CAPAエスカレーション
CAPAを上手に運用するための仕組み
11.CAPA7ステップ
Step 1 問題を検出し明確にする
Step 2 問題の原因を詳しく調査する
Step 3 根本原因の特定
Step 4 処置の特定と検証または妥当性確認
Step 5 予防または是正する為の処置
Step 6 処置の結果を文書化
Step 7 有効性の確認
12.SCARの実施
13.設計CAPA
14.CAPAマネジメント
マネジメントレビューの運用方法
15.品質問題の事例に学ぶ
Warning Letter Case Study
* 内容(項目)が変更される場合があります
2021_4_CAPA course
FDA 設計管理2日間HBコース Rev.27
〜 リスクをヘッジしコンプライアンスを堅持するフレームワーク 〜
NEW QMSR 対応 21CFR Part 820.30 / 75 ISO13485
QSR内部監査員コースご出席要件コースです
( オンサイト及び過去弊社コースを受講された方は要件を満たします )
会場参加、リモート、ハイブリッド形式で開催します
☆ 開催概要 ☆
ほとんどの日本企業に於ける医療機器開発・設計管理の仕組みは世界標準にほど遠く、これはグローバルに進出しようとする企業のみならず、患者の命を預かる全ての医療機器メーカーにとって大きなリスクを伴います。米国FDAは自らの査察で、医療機器品質不良の根本原因は上流側の設計管理にあると査察の結果から統計的に見抜いており、それはクオリス・イノーバの10年以上にわたるGAP監査でも明らかです。そこで本コースでは、グローバルな開発、設計管理を米国FDA「設計管理ガイダンス」及び欧米で取り入れられている開発フレームワークを元に体系的に解説、また、品質不良を発生させない仕組み(ロバスト設計)をタグチメソッドと設計CAPAを用いて解説します。 2日目はプロセスバリデーションの本質、工程内試験に頼らない均一で高品質な製品を一貫して製造する為、パラメータによる製造方法を実現する工程設計について解説します。 ”規制” だからではなく、設計品質を高めることにより患者さんの命を守り、企業にとっても大きな潜在的利益を得られるという大原則は、エンジニアの方々から多くの共感と支持を頂いてきたクオリス・イノーバの最も人気のある価値あるコースです。 このコースはオンサイトでも実施しております。お問い合わせはこちらから。
日 時: 2022年6月29日(水曜)、30日(木曜) 9:30〜16:30
締 切: 2022年6月22日(水曜)
会 場: 全国町村会館 大ホール(東京・永田町) 永田町駅から徒歩1分( 3番出口 )
リモートでのご参加はZOOMとなります
講 師: クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象: 医療機器・IVD市場にこれから参入しようとしている企業
医療機器・IVDを海外に輸出しようとしている企業
FDA査察準備、MDSAPを受審しようと準備している企業
既にFDA査察を終え、指摘対応に苦慮している企業
製品実現に関わる各組織( マーケティング、開発、生産技術、品証など )
受講料: ¥99,000( テキスト・昼食含む )
割 引: ¥88,000( テキスト・昼食含む )
1社2名様以上受講される場合
特 割: 再受講の方、プロジェクト終了企業様で継続研修としてご検討の方ご相談下さい。
定 員: クラス 定員 20名
リモート定員 30名
出席者全員に教育記録として利用できる修了書をお渡しします
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡いたします。
2名様以上ご参加の場合、お名前とローマ字読み、メールアドレスを連絡欄にご記入下さい。
持参品: 弊社QSR翻訳本第5版をご持参下さい。お持ちでない方はこちらからお求め頂けます。
ご注意: 本コースは、品質マネジメントシステムの一部を解説するため、システム全体の知識がないと理解できません。本コース受講前に5月開催QMSR2日間コースの受講をお勧めします。( QMSR2日間コースで訳本を配布しています )
開催要件:
1.コロナウイルス対応
行政の指示による規制の対象となった場合、リモートのみの開催となります。
弊社のウィルス対策を順守して戴きます。
2.リモートご参加要件
ZOOM以外での対応はしておりません
会社からご参加の場合、会議室から会社PCでのご参加となります。
自宅からご参加の場合、会社PCでのご参加となります。
その他、チェックリストでご確認頂く為、事前にご確認をお願いしています。
メールでお問い合わせ
電話でお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1日目(セミナー内容)
A.コンプライアンスとインテグリティー
設計不良の原因は何か
設計CAPAとは
ケーススタディ
不正を排除する仕組み
規制・規格に満足するればOKか?
人の命
行政によるミッションの相違
B.医療機器規制を理解する
新しい規則QMSRと今後の対応値
適用規制、規格、ガイダンス
1.Subpart C 設計管理 820.30 ISO13485 - 7.3
設計管理の目的とその重要性
プロセスアプローチの枠組と製品実現フレームワークの重要性
ライフサイクルマネジメント(LCM)と Waterfall Model
関連する ISO13485 の項目(プロセス)
Form 483 の FY 統計的解析から理解すること
2.820.30 (a) 総則 ISO13485 - 7.3.1
プロセスアプローチ ISO13485 4.1 の理解と
経営者の責任(リソースの提供とKPI)
製品実現プロセス
タートルチャート
リソースとしての力量(教育と訓練) 820.25
文書体系から俯瞰する設計管理プロセス
3.820.30 (b) 設計及び開発計画 ISO13485 - 7.3.2
医療機器開発ライフサイクルマネジメント(LCM)
設計品質確立の重要な要素; コンカレントエンジニアリング(CE)
PM、PLの役割と責任
ライフサイクルリスクマネジメントの理解(LCRM) ISO14971
タグチメソッド開発フレームワークによるフロントローディング手法
マーケティング戦略の重要性
開発計画書
4.820.30 (c) 設計インプット ISO13485 - 7.3.3
コミュニケーションエラー
曖昧なニーズの明確化
要求仕様から完成品までの一貫性
トレーサビリティーマトリックス(TMX)
ラベリング、包装設計のインプット
5.820.30 (d) 設計アウトプット ISO13485 - 7.3.4
タートルチャートのアウトプット
設計アウトプットのリスト
E−BOMの役割
製造仕様書とは
6.820.30 (e) 設計レビュー ISO13485 - 7.3.5
問題の早期発見、解決が目的でセレモニーで終わらせない仕組み
設計品質確立の重要な要素
設計レビューとテクニカルレビュー
レビューワーと責任と権限
7.820.30 (f) 設計検証 ISO13485 - 7.3.6
インプットからアウトプットまでのトレーサビリティー
製品の安全性、性能を証明するエビデンスの作り方
設計検証計画に必要な要素
設計検証レポートはデータの信頼性を確保
8.820.30 (g) 設計バリデーション ISO13485 - 7.3.7
設計検証と設計バリデーションの相違
設計バリデーションの手法
2つのバリデーションの相違と記録
ソフトウェアのバリデーションとVモデル
3種類のバリデーションの相違
9.820.30 (h) 設計移管 ISO13485 - 7.3.8 → 第2日目(工程設計)
10.820.30 (i ) 設計変更 ISO13485 - 7.3.9
変更管理プロセスに伴う設計変更
ライフサイクルリスクマネジメント
タートルチャート
設計変更フロー
11.820.30 (i ) 設計変更 ISO13485 - 7.3.10 設計・開発ファイル
DHF / DMR / DHR の定義
識別、ファイルの基本
文書体系と設計標準
2日目(セミナー内容)
設計移管と工程設計
820.30 (h)、13485 - 7.3.8、7.5.6
1.820.30 (h) 設計移管 ISO13485 - 7.3.8
ライフサイクルリスクマネジメントにおける設計移管(工程設計)
均一な製品を一貫して生産する手法
プロセスバリデーションの目的
2.なぜ工程バリデーションを実施するのか 820.75, ISO13485 - 7.5.6
プロセスバリデーションの目的とその本質
プロセスバリデーションの効果
工程管理、監視の目的と本質
医療機器規制・規格の理解
3.バリデーションすべき工程とは
バリデーションすべき工程の識別
検証工程とバリデーション工程の相違
設備バリデーションと工程バリデーション
バリデーション工程例
4.バリデーションの種類
バリデーションの種類
設備バリデーションと劣化パラメータ 820.70(g), 72(a)
コンピューターシステムバリデーション(CSV) 820.70(i), ISO13485 - 7.5.6 (g)
5.バリデーションステップ 820.75, ISO13485 - 7.5.6
VMP:バリデーションマスタープラン
DQ: 設計時適格性評価
IQ: 据付時適格性評価
OQ: 稼働時適格性評価
PQ: 製造時適格性評価
PPQ: プロセス街道性評価
バリデーション文書化
プロセスバリデーションの明確化
自動化工程の明確化
CSVカテゴリーの明確化
ソフトウェアのバリデーションの基本
リスクベースCSV
6.工程設計とプロセスバリデーション 820.30 (h)、 ISO13485 - 7.3.8
ライフサイクルマネジメントに於ける工程設計
工程設計の進め方
工程設計とプロセスリスク分析( PFMEA )
環境管理 820.70(c)、 ISO13485 - 6.4
汚染管理 820.70(e)、 ISO13485 - 6.4
副資材 820.70(h)、 ISO13485 - 7.5.2
工程設計PTMX
QC工程表
7.回顧的バリデーション
8.バリデーションのアウトプット
QC工程表と統計的手法(工程能力)
リバリデーション設定
設備インスペクションとメンテナンス
( 内容が変更される場合がございます )

2021_11 設計管理2日間コース
QMS 購買管理HBコース
〜 SCM でQCDに貢献する購買プロセスの本質 〜
21CFR Part 820・ISO13485・ISO9001
このコースの売り上げ金の一部は人道支援活動に寄付されます
会場参加、リモート、ハイブリッド形式で開催されます
弊社コロナウィルス対策はこちらからご確認下さい
☆ 開催概要 ☆
購買プロセスは言われた通りにモノを買っていればよいというものではなく、サプライチェーン全体を俯瞰し、製品企画時の上流から積極的にコンカレントで参画、最も効率よいQCDを提案することこそ会社に貢献するプロセスとして価値があります。その為には、戦略的な組織体制とフレームを構築する必要があり、開発購買や情報購買と言ったフレームワークは早期の製品ローンチを実現します。また、アッセンブルラーが多くなった昨今、行政当局も購買プロセスに多くの査察時間を費やすようになりました。自社と同じシステムで管理する必要性が高まり、フレームと管理能力が要求されます。FDA QSRのみならず、ISOの基本9001の要求も加味しながら理解を深めます。
オンサイトでも可能です。お問い合わせはこちらから。
日 時:2022年6月6日(月曜) 9:30 〜 16:30
会 場:全国町村会館(東京・永田町) 永田町駅から徒歩1分( 3番出口 )
リモートでご参加なさる方は ZOOM でのご参加となります
講 師:クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
ODM、OEMとして設計・製造を受託している企業
受講料: ¥55.000-( 消費税込、テキスト、ご来場の方には昼食を提供 )
リモートでご参加なさる方には、事前にテキスト等を郵送致します
割 引: ¥45.000-( 消費税込、テキスト、ご来場の方には昼食を提供 )
1社2名様以上ご参加の場合
定 員: クラス 定員 20名
リモート定員 30名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
開催要件:
1.リモート参加要件
会社からの場合、個室(会議室等)から会社PCで御参加となります。
ご自宅からの場合、会社PCでのご参加となります。
その他チェックリストを事前にお渡しし環境をご確認頂きます。
2.コロナウィルス対応
弊社のウィルス対策を順守して頂きます。
当日発熱がある場合、参加を御控え頂きます。
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
A.FDA規制、査察の最新情報と傾向分析(QMSRの動向)
1.適用規制・規格
FDA QSR,ISO13485,9001の要求とは
プロセスアプローチの購買プロセスとは
2.購買管理の目的
規格が示す購買管理の目的
製品、サービス購買とは
4つの購買範囲とは
3.用語の定義
製品購買、サービス購買とは
プロバイダー、サプライヤー、コントラクターとは
4.購買管理
GHTF( IMDRF )ガイダンスのフロー
5.購買方針
購買方針がなければ購買プロセスは成り立たない
6.戦略・開発購買
サプライチェーン全体を俯瞰した戦略
開発スピードを上げ原価削減、購買活動のスピードを上げある
ベストプラクティスの収集と技術の取り込み
7.購買計画
製品企画段階から参画
供給する製品、サービスの特定
技術及びプロセス情報の特定
供給者候補の特定
リスク、管理方法の特定
8.供給者候補選定
供給者ビジネス、運用能力評価
供給者候補選定
9.供給者選定
評価選定基準の策定
供給者候補の能力評価
供給者の承認
10.管理方法の取り決め
購買情報とは
管理方法
11.変更時対応
変更管理規定
4Mと変化点管理
12.納期、測定、監視
製品、サービスの検証
不適合の手順
供給者の監視
製品の受領、出荷場、冷蔵室、NCエリア
カーゴ内温度バリデーション
13.フィードバック、コミュニケーション
CR
SCAR
供給者評価シート
14.サプライヤー監査
リスクベース監査
5つの監査、評価エリア
情報購買、戦略上重要な監査チーム
( 内容が変更になる場合もございますが大きな変更はございません。)

2021_11_PC
QMSR ショートセッション
QSRに変わる新しい規制の解説
☆ 開催概要 ☆
FDAが3月2日に実施したQSRに変わる新しい規制QMSRのカンファレンスの内容について概要を説明するショートセッションです。リモートのみでの開催となります。
1.QMSRのロードマップ
2.CFR Part 820 の扱い
3.QSRとQMSR、ISO13485の相違
4.今後のFDA査察
日 時: 1.4月15日 15時〜16時 ( メーリングリストでメール受領者限定 )
2.4月28日 15時〜16時
会 場: リモートのみ( ZOOM )
講 師: クオリス・イノーバ株式会社
代表取締役社長 木村 浩実
受講料: 無料
定 員: いずれも80名様限定
お申込: 4月28日のみお申し込みできます。
下記よりお申込ください。追ってZOOMのリンクをお知らせします。
ご注意: ZOOM以外では実施しておりません。
氏名表示し、カメラで参加者が確認できる必要があります。
氏名やカメラ映像は他の参加者には映らないモードで実施します。
メールでのお問い合わせ
FDA QMSR/QSR/13485 2日間HBコース
〜 医療機器・IVDに求められるQMSの本質を学ぶ 〜
21CFR Part 820
このコースは、オンサイト、リモートのハイブリッド開催です。
2022年3月に公開された QSR に変わる新たな規制 QMSR について、
ISO13485 との相違はもとより、原点に立ち返る QMS の本質について学ぶ新たなコースです。
年末に実施する監査員コースの受講要件となります。
☆ 開催概要 ☆
QSR翻訳本(第5版)+QMSR翻訳本(第1版)付き
米国が最も大きなマーケットであることは医療機器でも例外では無く、米国での成功がグローバルを制すると言っても過言ではありません。しかし、米国で製品を上市するには医療機器規制 QMSR に基づく QMS の構築は必須となります。2023年から新たに適用となるこの規制は、FDA QSR 規制を積極的に取り入れてきた ISO13485(2016) にほぼ準拠となりますが、ガイダンスなどは今まで通り適用となります。今後は QSR の欠点でもあった QMS の本質、リスクベースプロセスアプローチのフレームワークが必須となります。弊社の経験上 ISO1345 取得済みの企業であっても ISO に準拠しているとはとても言いがたい状況で QMSR には準拠できませんのでご注意下さい。今まで弊社は一貫して規制の本質を解き、事業活動とのインテグレーションを取ることにより、結果の出せるマネジメントシステムを提唱し、多くの企業の皆様の共感を集め支持されて参りました。米国に進出する企業のみならず、医療機器・IVDを開発しようとしている企業の皆様のマインドが大きく変わる価値あるクオリス・イノーバのメインコースです。
新規制が適用となるまで QSR が適用となる為、本コースでは現行 QSR, QSMR, ISO13485 について、リスクベースプロセスアプローチ、製品実現フレームワークといったシステムの根幹となるフレームワークを中心に概要を説明します。
オンサイト(一定人数が集まった会社でのセミナー)も可能です。お問合せはこちらから。
日 時: 2022年5月25日(水曜)、26日(木曜) 9:30〜16:30
会 場: 全国町村会館(東京・永田町)(永田町駅から徒歩1分)
リモートで参加なさる場合、ZOOMでの御参加となります。
講 師: クオリス・イノーバ株式会社
取締役社長 木村 浩実
対 象: 医療機器・IVD市場にこれから参入しようと考えている企業
米国に輸出を計画している企業
米国FDA 査察準備を考えている企業
ODM、OEM として医療機器を受託しようとしている企業
受講料: ¥99,000−(税込)( 2日分、テキスト、対訳本2冊、昼食代含 )
リモートで参加なさる方には事前にテキスト、対訳本を郵送いたします。
割 引: ¥88,000−(税込)
1社2名以上ご参加の場合
特 割: 企業で行うQMS構築プロジェクトなどや企業で弊社が実施した継続研修などで
ご利用の場合、特別割引を適用させて頂きますのでメールでご相談下さい。
満席となった場合、正規料金をお支払いの方が優先となりご希望に添えない場合
がございますので予めご了承ください。
定 員: クラス 定員 20名様 5月18日締切
リモート定員 30名様
( 出席者全員に教育記録として利用できる修了書をお渡しします )
開催要件:
1.リモート参加要件
会社からの場合、個室(会議室等)から会社PCで御参加となります。
ご自宅からの場合、会社PCでのご参加となります。
その他チェックリストを事前にお渡しし環境をご確認頂きます。
2.コロナウィルス対応
弊社のウィルス対策を順守して頂きます。
当日発熱がある場合、参加を御控え頂きます。
お申込: セミナー申込みページからお申し込み下さい。
折り返し、会場、お振り込み先情報などをメールでご案内致します。
2名以上で参加なさる方ははその旨ご記入下さい。
割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.コンプライアンスとインテグリティー
患者の命と会社の危機管理
データインテグリティー
2.医療機器規制の理解
行政によるミッションの相違
米国規制法
医療機器規制の理解
3.QSR から QMSR へ移行する経緯
4.QSR と QMSR 、ISO13485( 2016) との相違
5.QMSR 移行後変わるものと変わらないもの
6.なぜ ISO 認証では FDA 査察にパスしないのか
7.QSIT アプローチとは
8.QMSR
Subpart A 総則
820.1 適用範囲
820.3 定義
規制を正しく理解する要素:定義
820.7 参照による引用
規制に記載していない ISO の引用について
820.10 品質マネジメントシステムに関する要求事項
ISO13485(2016) 適用の要件
820.15 コンセプトの明確化
Subpart B 附則
820.35 記録の管理
820.45 機器のラベリング及び包装管理
9.QMS を構成するフレームワーク
QSR ISO13485 (2016)
A. リスクベースプロセスアプローチ
4. 品質マネジメントシステム
820.20 経営者の責任 5. 経営者の責任
820.22 品質監査 8.2.4 内部監査
820.25 要員 6. 資源の運用管理
820.40 文書管理 4.2 文書化に関する要求事項
820.90 不適合製品 8.3 不適合製品の管理
820.100 是正処置及び予防処置 8.5 改善
820.250 統計的手法 8 測定、分析及び改善
B.製品実現フレームワーク
820.30 設計管理 7.3 設計・開発
820.50 購買管理 7.4 購買
820.60 識別 7.5.8 識別
820.65 トレーサビリティー 7.5.9 トレーサビリティー
820.70 生産及びプロセスの管理 7.5 製造及びサービスの提供
820.72 検査、測定及び検査装置 7.6 監視機器及び測定機器の管理
820.75 プロセスバリデーション 7.5.6 プロセスのバリデーション
820.80 受入、工程内及び完成機器のアクセプタンス
7.4.3 購買製品の検証
820.86 アクセプタンス状態 7.5.8 識別
820.120 機器のラベリング
820.130 機器の包装
820.140 取扱い 7.5.11 製品の保存
820.150 保管 7.5.11 製品の保存
820.160 流通 7.5.11 製品の保存
820.170 据付け 7.5.3 据付け活動
820.180 (記録) 4.2.5 記録の管理
820.181 DMR 4.2.3 医療機器ファイル
820.184 DHR 7.1 製品実現の計画
820.186 QSR 4.2.4 文書管理
820.198 苦情ファイル 8.2.2 苦情ファイル
820.200 附帯サービス 7.5.4 附帯サービス活動
2022-10 QSR seminar

FDA MDR&CAR コース Rev.8
リモート参加も可能なハイブリッド形式
〜 指摘の多い有害事象報告・リコール規制の理解とプロセス構築 〜
21CFR : Part 820, 803, 806
このコースの売り上げ金の一部は人道支援活動に寄付されます
☆ 開催概要 ☆
FDA査察レポートの解析からワースト4位(全体の約10%)に急浮上しているMDR(有害事象報告)、日本でも Warning Letter (警告書)を受けているほとんどの企業がMDRの指摘を受けています。昨今のデータ偽装事件を受け、FDAはこれらにフォーカスし厳しく査察を行っています。( 2021年レポート)。これはMDR報告の判断に迷ったら報告するという米国の文化とは異なり、日本のメーカーは情報を積極的に発信せず隠すという古い会社の文化が根強くあることが要因の一つですが、そもそも規制を理解せず正しい判断を行っていないことが大きな原因です。医療機器を作っているメーカーは、患者さんの命を守るという社会的な責任があります。会社を守っているようでも、結果的に会社が倒産に追い込まれることもある非常にリスクの高いプロセスであることを理解すべきです。それには規制の正しい理解、規制を守る仕組みと組織が必要です。このセミナーでは、CAPAサブシステム(苦情、不適合、CAPA)の各プロセスとリンクし、日本のメーカーの Warning Letter (警告書)を引き合いに正しいMDRプロセスと関連するCAR(リコール)プロセスを構築することを理解します。今回は薬機法のGVP規制とのリンクも解説します。
このコースはオンサイトでも実施しています。お問い合わせはこちらから。
日 時:2021年3月7日(月曜)10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
リモートはZOOMから
対 象:既に医療機器・IVDを米国に輸出している企業
これから医療機器・IVDを米国輸出する準備を行っている企業
既にFDA査察を終え対応に困っている企業
受講料: ¥55,000-( 消費税込み、テキスト代含む、ご来場の方には昼食代含まれます )
割 引: ¥44,000-( 消費税込み、テキスト代含む、ご来場の方には昼食代含まれます )
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
特 割:弊社プロジェクトを実施中、又は継続研修をご希望の場合、御負担軽減のためさらに
割引をさせて頂きますのでお問い合わせください。この場合、正規料金でお申込の方
が優先され、出席できない場合がございますので予めご了承ください。
再受講:再受講をご希望の方はさらに割引が適用され、空席がある場合ご出席頂けます。
併せて過去ご出席なさった日をお知らせ下さい。
お申込:セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
リモートをご希望の方はその旨ご記入下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.規制を理解する
事例と統計からみる規制違反
理解すべき規制・規格・ガイダンス
eMDR規制
苦情組織と力量
規制が求めるあるべき組織と力量
2.有害事象報告( MDR )Part 803
MDR規制の基本要件
FDA Warning letter から
規制の目的、重要性
MDRとして報告すべき事象とは
2.1 定義を理解する
重篤な障害
機能不全
報告すべき Malfunction
合理的に推測する
認識する
2.2 4.5日、30日報告
報告形態と時間軸
5日報告
ディシジョンツリー
2.3 FDAへの報告
報告フロー
報告様式 3500A
報告不要の場合
報告書の種類
報告免除
Warning letter 警告書
2.4 MDRプロセス
MDR報告フロー
CA,PAプロセス
3.回収報告(CAR)Part 806
規制の理解
報告免除
定義
リコールフロー
任意と強制リコール
定期メンテナンスとリコール
3.1 リコール判断基準
リコール判断基準例
ラベリングの定義
ラベリング違反と法的根拠
報告すべきラベリング違反例
3.2 CAR ディシジョンツリー
リコールディシジョンツリー
3.3 報告書と処理フロー
リコール判断後フロー
リコールクラス
有効性の確認
CAPAとの関係
4.警告書から理解すべき注意点とプロセスへの展開
Warning Letter ( 警告書 )の事例から理解する
警告書から理解すべき注意点とプロセスへの展開
プロセス構築後のレトロスペクティブな是正とは
*コース内容が一部変更される場合がございますが大きな変更はございません。

2021_3_MDR/CAR
〜 指摘の最も多いCAPAの理解と問題の予防、再発防止の仕組み 〜
このコースの売上金の一部は人道支援に寄付されます
* コース概要 *
CAPAプロセスを確認するだけで、その会社の品質システムや品質に対する理解度がよくわかります。CAPAプロセスに問題があれば、品質マネジメントシステムの適切性、妥当性はなく、特にマネジメントレビューがまともに運用されているとは到底思われません。品質マネジメントシステムの原則はプロセスアプローチ、PDCAをまわす為、マネジメントレビューのインプットでプロセスを監視し、改善ツールであるCAPAでプロセスを改善します。当たり前のことがシステムとして運用されていないことを理解している規制当局は、CAPAプロセスにフォーカスして確認しようとします。QMSが機能していないことは昨今の品質に関わる事件から明らかで、医療機器・IVDの場合、命に関わる大問題です。本コースでは今までの内容を大きく変更し、プロセスアプローチの手法に重点を置き、事例を元にCAPAの本質を理解して頂きます。 このコースは受講者と同じ目線の御社事例を用いることでより理解が高まるオンサイトセミナーが可能です。お問合せはこちらから。
日 時:2022年1月17日(月曜)10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 ) ( 永田町駅から徒歩1分 )
講 師:クオリス・イノーバ株式会社代表取締役社長 木村 浩実
対 象:医療機器、IVD、医薬品等に限定せず、CAPAプロセスを学びたい企業
受講料: ¥55,000 消費税込( テキスト代・昼食含む(ご来場の方のみ) )
割 引: ¥44,000 消費税込( テキスト代・昼食含む(ご来場の方のみ) )
1社2名様以上ご参加の場合
特 割:弊社プロジェクトを実施中の場合や、継続研修をご希望の場合、ご負担軽減のためさらに
割引をさせて頂きますのでお問い合わせください。 この場合、正規料金をお支払いのお
客様が優先され、ご出席できない場合がございますのでご了解ください。
再受講:空席がある場合、半額で再受講可能ですのでお問い合わせください。
併せて受講された日もご記入ください。
定 員:20名様 1月12日締め切り
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
開催要件;コロナウィルスの対応について
1.政府の緊急事態宣言が発令された場合延期開催となります
2.お一人様1テーブルのご使用となります
3.換気は自動換気システムが作動し入口の扉を開けて開催します
4.入口にアルコールを設置、手指消毒と服、靴ウラの消毒をお願いします
5.テーブル、席はホテル側で消毒が行われます
6.講師、スタッフ、出席者全員マスク着用でお願いいたします
お申込:セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールで連絡いたします。
複数名ご参加の場合、同様の情報をご記入下さい。 割引料金でご案内いたします。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.なぜFDAはCAPAに注目するのか
2.なぜ品質問題は再発するのか
3.規制・規格を正しく理解する
4.プロセスアプローチの基本
プロセス関連図
タートルチャート
5.TQMの仕組みが不可欠
マネジメントレビュー
PDCAをまわす
プロセス改善ツール
6.用語の正しい理解
修正、是正、予防の相違
逸脱、不適合、逸脱許可と特別採用の相違
修正、手直し、修理の相違
7.CAPAプロセスの IN・OUT
CAPAタートルチャート
不適合処理プロセス
苦情処理プロセス
CAPAプロセス
MDRプロセス
リコールプロセス
8.直接原因と根本原因を理解する
直接原因と根本原因の相違
根本原因の改善がプロセスアプローチの本質
9.予防プロセス
品質管理の原則、予防活動の理解
10.エスカレーション
CAPAエスカレーション
CAPAを上手に運用するための仕組み
11.CAPA7ステップ
Step 1 問題を検出し明確にする
Step 2 問題の原因を詳しく調査する
Step 3 根本原因の特定
Step 4 処置の特定と検証または妥当性確認
Step 5 予防または是正する為の処置
Step 6 処置の結果を文書化
Step 7 有効性の確認
12.SCARの実施
13.設計CAPA
14.CAPAマネジメント
マネジメントレビューの運用方法
15.品質問題の事例に学ぶ
Warning Letter Case Study
* 内容(項目)が変更される場合があります
2021_4_CAPA course
FDA 設計管理2日間コース Rev.25
〜 リスクをヘッジしコンプライアンスを堅持するフレームワーク 〜
21CFR Part 820.30 / 75 ISO13485 7.3 / 7.5.6
QSR内部監査員コースご出席要件コースです
( オンサイト及び過去弊社コースを受講された方は要件を満たします )
条件付きではございますが、リモートでも参加頂けます
会場参加、リモート、ハイブリッド形式で開催します
☆ 開催概要 ☆
ほとんどの日本企業に於ける医療機器開発・設計管理の仕組みは世界標準にほど遠く、これはグローバルに進出しようとする企業のみならず、患者の命を預かる全ての医療機器メーカーにとって大きなリスクを伴います。米国FDAは自らの査察で、医療機器品質不良の根本原因は上流側の設計管理にあると査察の結果から統計的に見抜いており、それはクオリス・イノーバの10年以上にわたるGAP監査でも明らかです。そこで本コースでは、グローバルな開発、設計管理を米国FDA「設計管理ガイダンス」及び欧米で取り入れられている開発フレームワークを元に体系的に解説、また、品質不良を発生させない仕組み(ロバスト設計)をタグチメソッドと設計CAPAを用いて解説します。 2日目はプロセスバリデーションの本質、工程内試験に頼らない均一で高品質な製品を一貫して製造する為、パラメータによる製造方法を実現する工程設計について解説します。 ”規制” だからではなく、設計品質を高めることにより患者さんの命を守り、企業にとっても大きな潜在的利益を得られるという大原則は、エンジニアの方々から多くの共感と支持を頂いてきたクオリス・イノーバの最も人気のある価値あるコースです。 このコースはオンサイトでも実施しております。お問い合わせはこちらから。
日 時: 2021年11月29日(月曜)、30日(火曜) 10:00〜16:30
会 場: 全国町村会館 大ホール(東京・永田町) 永田町駅から徒歩1分( 3番出口 )
リモートでのご参加はZOOMとなります
講 師: クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象: 医療機器・IVD市場にこれから参入しようとしている企業
医療機器・IVDを海外に輸出しようとしている企業
FDA査察準備、MDSAPを受審しようと準備している企業
既にFDA査察を終え、指摘対応に苦慮している企業
受講料: ¥99,000( テキスト・昼食含む )
割 引: ¥85,000( テキスト・昼食含む )
1社2名様以上受講される場合
定 員: 24名
出席者全員に教育記録として利用できる修了書をお渡しします
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡いたします。
2名様以上ご参加の場合、お名前とローマ字読み、メールアドレスを連絡欄にご記入下さい。
持参品: 弊社QSR翻訳本第5版をご持参下さい。お持ちでない方はこちらからお求め頂けます。
ご注意: 本コースは、品質システムの一部を解説するため、システム全体の知識がないと理解できません。本コース受講前に10月開催QSR2日間コースの受講をお勧めします。( QSR2日間コースで
訳本を配布しています )リモートでのご参加希望が叶えられない場合もございます。
メールでお問い合わせ
電話でお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1日目(セミナー内容)
A.設計不良はなぜ発生し、再発するのか?
ケーススタディー
直接原因と根本原因は異なる
繰り返される設計品質に起因する製品不良
ISOとQSRの本質的な相違
規制・規格に満足すればOKか?
理解すべき規制・規格・ガイダンス
設計品質の本質的価値
1.Subpart C 設計管理
Waterfall Model と 820.30
ISO13485 vs FDA QSR
FY 483 解析から理解すること
2.820.30 (a) 総則
手順を確立し維持するとは
重要な品質要素、規制要求事項
経営者が提供する設計環境
経営者の責任
プロセスアプローチに基づくリスクヘッジと品質計画
タートルチャートによるリソースとKPIの見定め
リソースとしての要員( 820.25 )
教育と訓練( 訓練の重要性 )
文書体系の基本
3.820.30 (b) 設計及び開発計画
医療機器開発ライフサイクルマネジメント(LCM)
設計品質確立の重要な要素、コンカレントエンジニアリング(CE)
PM、PLの役割
ライフサイクルマネジメント・リスクマネジメント
タグチメソッドの開発方式によるフロントローディング手法
カスタマーニーズを要求仕様に変換するQFD
設計計画書とは
4.820.30 (c) 設計インプット
コミュニケーションエラー
要求仕様書から設計仕様書への変換
曖昧なニーズのスペックへの変換
重要な設計要素トレーサビリティーマトリックス(TMX)
ラベリング、包装設計のインプット
5.820.30 (d) 設計アウトプット
入力のバラツキを押さえたアウトプットの確認
設計アウトプットを定義しアウトプット文書を明確にする
E-BOM の役割
製造仕様書とは
図面発行
DMRとトレーサビリティー
6.820.30 (e) 設計レビュー
問題の早期発見、解決が目的でセレモニーで終わらせない
設計審査で設計のロバストネスを確認
設計審査とテクニカルレビュー
審査者の力量
7.820.30 (f) 設計検証
設計検証と設計バリデーションの相違
インプットからアウトプットまでのトレーサビリティー
エビデンスに基づく設計
設計検証計画に必要な要素
設計検証レポートはデータの信頼性を確保
8.820.30 (g) 設計バリデーション
設計バリデーションの手法
社内、臨床評価の相違と記録
ソフトウェアのバリデーションとVモデル
3種類のバリデーションの相違
9.820.30 (h) 設計移管 → 第2日目(工程設計)
10.820.30 (i ) 設計変更
DHF / DMR / DHR の定義
識別、ファイルの基本
文書体系と設計標準
2日目(セミナー内容)
設計移管と工程設計
1.なぜ工程バリデーションを実施するのか
プロセスバリデーションの目的とその本質
プロセスバリデーションの効果
工程管理、監視の目的と本質
医療機器規制・規格の理解
2.バリデーションすべき工程とは
バリデーションすべき工程の識別
検証工程とバリデーション工程の相違
設備バリデーションと工程バリデーション
バリデーション工程例
3.バリデーションの種類
バリデーションの種類
設備バリデーションと劣化パラメータ
コンピューターシステムバリデーション(CSV)
4.工程設計とバリデーションステップ
VMP:バリデーションマスタープラン
DQ: 設計時適格性評価
IQ: 据付時適格性評価
OQ: 稼働時適格性評価
PQ: 製造時適格性評価
PPQ: プロセス街道性評価
バリデーション文書化
プロセスバリデーションの明確化
自動化工程の明確化
CSVカテゴリーの明確化
ソフトウェアのバリデーションの基本
リスクベースCSV
5.820.30 (h) 設計移管
820.75 プロセスバリデーション
820.72 検査、測定および試験装置
設計移管とプロセスバリデーションの関係
EV、PV、検査測定装置の妥当性とは
ミニスケールとフルスケール
工程設計とPTMX
6.バリデーションの実践
バリデーションマスタープラン(VMP)
バリデーション工程の識別
バリデーション計画
設備リストとバリデーション工程リスト
チームビルディング
DQP/DQR
IQP/IQR
OQP/OQR
PQP/PQR
PPQP/PPQR
7.バリデーションアウトプット
工程能力
再バリデーション設定
設備メンテナンス手順
( 内容が変更される場合がございます )

2021_6 設計管理2日間コース
FDA QSR2日間コース Rev.26
〜 医療機器・IVDに求められる規制要求事項の本質を学ぶ 〜
21CFR Part 820
このコースの売り上げ金の一部は人道支援活動に寄付されます
ISO13485 との比較をより詳細に行っておりシステム構築に最適です
設計管理コース、CAPAコースを受講される方も本コースを受講されることをお勧めします
内部監査員コース受講の要件こーすです
このコースは9月開催から延期となり、
条件付きではございますがウェビナーでもご参加頂ける
会場参加、リモート、ハイブリッド形式となります。
☆ 開催概要 ☆
* FDA 品質システム規制 QSR 対訳・解説本( 第5版 )付き
グローバルに進出しようとする企業にとって 米国は最も大きなマーケットであり、米国市場での成功がグローバルを制するといっても過言ではありません。しかし、米国に上市するには医療機器規制QSRに基づくFDAの査察は避けられません。( MDSAPを受審される企業も必須 )また、医療機器を国内で販売する際にもFDA QSRのシステムフレームワーク、エビデンスベースの考え方を学び取り入れる事は、製品品質確保につながり、リスクの低減、新しいISO13485にも準拠し、特に受託設計、製造会社 ( ODM、OEM ) では会社のプレゼンスを高め、サプライヤ−としての評価を高めます。 クオリス・イノーバのQSRコースは、多くのFDA査察準備、対応の経験や品質管理の経験から、FDA規制のみならず医療機器開発・製造の本質を鋭く説き、多くの企業の皆様の共感を集め支持されて参りました。米国に進出する企業のみならず、医療機器・IVD を開発しようとしている企業の皆様のマインドが大きく変わる価値あるクオリス・イノーバのメインコースです。
オンサイトでも可能です。お問い合わせはこちらから。
日 時:2021年10月18日(月)、19日(火) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
リモートでご参加なさる方は ZOOM でのご参加となります
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
ODM、OEMとして設計・製造を受託している企業
受講料: ¥99,000( 2日分、テキスト・対訳・解説本(5版)・昼食代含む )
リモートでご参加なさる方には、事前にテキスト等を郵送致します
割 引: ¥85,000
1社2名様以上ご参加の場合
特 割: 企業で行うプロジェクトの後、継続研修などでご利用の場合、
特別割引を適用させて頂きますのでメールでご相談下さい。
この場合、正規料金をお支払いの方が優先となりご希望に添えない場合が
ございますので予めご了承ください。
定 員: 20名 9月15日締切
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
開催要件;コロナウィルスの対応について
1.換気は自動換気システムが作動し、入口の扉を開けて開催します
2.入口にアルコールを設置、手指消毒と服、靴ウラの消毒をお願いします
3.テーブル、席はホテル側で消毒が行われます。
4.講師、スタッフ、出席者全員マスク着用でお願いいたします
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
第1日目 セミナー内容
1.コンプライアンスとインテグリティー
患者の命と会社の危機管理
データインテグリティー
トップのリーダーシップ
2.行政によるミッションの相違
3.米国法規制の理解
4.医療機器規制の理解
5.なぜISOではFDA査察にパスしないのか
6.QSITアプローチとは
7.規制を正しく理解する要素:定義
820.20 経営者の責任
経営者がリードするTQMが基本
マネジメントレビューの本質、プロセスアプローチ
プロセスを常に改善する
820.22 品質監査
効果的な監査手法と監査報告のあり方
内部監査はプロセスの異常を見極める
内部監査員はなぜ異常を見極められないのか
820.25 要員
教育の重要性:教育の手法と記録、力量表、リソースとは
教育と訓練の相違、アウェアネストレーニングの実施と訓練の重要性
820.30 設計管理( 概要のみ:10月開催 設計監理2日コースで詳細解説 )
ライフサイクル製品開発手法、ウオーターフォールモデル、
フロントローディング手法、ロバスト設計
設計検証と設計バリデーションの相違
今までISO になかった設計移管、工程設計とは
設計CAPAとは
820.40 文書管理
グロバールに通用する文書管理
品質を担保するSOPの作成方法
820.50 購買管理
開発購買とその役割、購買プロセス
3購買(製品、サービス、一般)と購買製品検証
新規サプライヤ評価と継続評価
購買情報とは
( 概要のみ:9月開催 購買管理コースで詳細解説 )
820.60 識別
混同を防止する為の識別方法とは
820.65 トレーサビリティー
体内埋込型機器だけではなく、変化点を知り品質を高める
820.80 アクセプタンスアクティビティー
3つのアクセプタンスを理解する
Receiving, In-process, and Finished device acceptance
820.86 アクセプタンス状態
状態表示する意味とは
820.90 不適合品
不適合品の処理プロセス
FDAはなぜNCを見たがるのか
グローバル用語を使用し、CAPAとのリンクを示す
820.120 機器のラベリング
リコールが一番多く査察で注目されるラベリングの出納管理
ラベリングの設計とは
ラベルとラベリングの相違
820.130 機器の梱包
梱包箱を見ればどんな設計を行っていたかが判る
設計依託先はISOで十分か?
820.140 取扱い
混同・劣化・損傷・汚染を発生させないためのリスク管理
820.150 保管
リスクベース保管管理
820.160 流通
ユーザーに届けるまでの製品設計
リスクベース流通管理
820.170 据付け
インスタレーション手順書のV&V
820.180 記録
全ての活動が根拠に基づくエビデンスで保証されるための重要な活動
Objective Evidence を示すための記録管理とは
820.181 機器原簿(DMR)
DMRを定義し識別する
DHF,DMR,DHRの相違
820.184 機器履歴簿(DHR)
製造記録の記載方法
コンプライアンス、製品品質を立証するため重要なDHR
820.186 品質システム記録
DHF,DMR,DHR以外の品質記録とは
820.200 付帯サービス
サービス記録の分析とMR報告,MDRの判定
820.250 統計的手法
TQMを進めていく上で最も重要な要素
品質改善に欠かせない統計的手法
第2日目 セミナー内容
820.70 生産及び工程管理
工程設計と工程V&V
( 概要のみ:10月開催 設計監理2日コースで詳細解説 )
工程のリスクマネジメント(PFMEA)
工程を監視、管理する工程パラメータとは
要員、環境、汚染管理
設備の妥当性とは
保守と検査の相違
副資材の定義と管理
820.72 検査・測定及び試験装置
意図した用途への適合性、測定器の精度管理とは
測定器の精度管理と救済処置
820.75 工程の妥当性確認
QSR,医療機器規制の本質、工程バリデーションとは
( 概要のみ:10月開催 設計監理2日コースで詳細解説 )
TQMが目指すべき試験レスとは
バリデーションの手法( VMP,DQ、IQ、OQ、PQ、PPQ )
設備バリデーションとは
CSV( コンピュータシステムバリデーション )とは
820.198 苦情ファイル
査察のキーポイント、苦情管理の目的を理解する
苦情処理とMDR( Part 803 )、CAR( Part 806 ) の関わり
( 概要のみ:MDR/CARコースで詳細解説 )
820.100 是正処置及び予防処置( CAPA )
FDA警告書で最も多い指摘CAPAの原則を理解する
品質システムの本質、プロセスアプローチの重要な要素
修正、是正、予防の相違を正確に理解しISOマインドを変える
CAPA7ステップを理解する
CAPAの効果(再発防止)を出す仕組み
MRで監視すべきCAPAプロセス
( 概要のみ:CAPAコースで詳細解説 )
( 内容が変更になる場合もございますが大きな変更はございません。)
20201_5 QSR Seminar 様子

QMS 購買管理コース
〜 SCM でQCDに貢献する購買プロセスの本質 〜
21CFR Part 820・ISO13485・ISO9001
このコースの売り上げ金の一部は人道支援活動に寄付されます
弊社コロナウィルス対策はこちらからご確認下さい
このコースは9月開催から延期となり、
条件付きではございますがウェビナーでもご参加頂ける
会場参加、リモート、ハイブリッド形式となります。
☆ 開催概要 ☆
購買プロセスは言われた通りにモノを買っていればよいというものではなく、サプライチェーン全体を俯瞰し、製品企画時の上流から積極的にコンカレントで参画、最も効率よいQCDを提案することこそ会社に貢献するプロセスとして価値があります。その為には、戦略的な組織体制とフレームを構築する必要があり、開発購買や情報購買と言ったフレームワークは早期の製品ローンチを実現します。また、アッセンブルラーが多くなった昨今、行政当局も購買プロセスに多くの査察時間を費やすようになりました。自社と同じシステムで管理する必要性が高まり、フレームと管理能力が要求されます。FDA QSRのみならず、ISOの基本9001の要求も加味しながら理解を深めます。
オンサイトでも可能です。お問い合わせはこちらから。
日 時:2021年11月22日(月曜) 10:00 〜 16:00
会 場:全国町村会館(東京・永田町) 永田町駅から徒歩1分( 3番出口 )
リモートでご参加なさる方は ZOOM でのご参加となります
講 師:クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
ODM、OEMとして設計・製造を受託している企業
受講料: ¥50.000-( テキスト代・昼食含む )
リモートでご参加なさる方には、事前にテキスト等を郵送致します
割 引: ¥40.000-( 1社2名様以上ご参加の場合 )
定 員: 20名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
A.FDA規制、査察の最新情報と傾向分析
1.適用規制・規格
FDA QSR,ISO13485,9001の要求とは
プロセスアプローチの購買プロセスとは
2.購買管理の目的
規格が示す購買管理の目的
製品、サービス購買とは
4つの購買範囲とは
3.用語の定義
製品購買、サービス購買とは
プロバイダー、サプライヤー、コントラクターとは
4.購買管理
GHTF( IMDRF )ガイダンスのフロー
5.購買方針
購買方針がなければ購買プロセスは成り立たない
6.戦略・開発購買
サプライチェーン全体を俯瞰した戦略
開発スピードを上げ原価削減、購買活動のスピードを上げある
ベストプラクティスの収集と技術の取り込み
7.購買計画
製品企画段階から参画
供給する製品、サービスの特定
技術及びプロセス情報の特定
供給者候補の特定
リスク、管理方法の特定
8.供給者候補選定
供給者ビジネス、運用能力評価
供給者候補選定
9.供給者選定
評価選定基準の策定
供給者候補の能力評価
供給者の承認
10.管理方法の取り決め
購買情報とは
管理方法
11.変更時対応
変更管理規定
4Mと変化点管理
12.納期、測定、監視
製品、サービスの検証
不適合の手順
供給者の監視
製品の受領、出荷場、冷蔵室、NCエリア
カーゴ内温度バリデーション
13.フィードバック、コミュニケーション
CR
SCAR
供給者評価シート
14.サプライヤー監査
リスクベース監査
5つの監査、評価エリア
情報購買、戦略上重要な監査チーム
( 内容が変更になる場合もございますが大きな変更はございません。)

2019_07_PC
FDA 設計管理2日間コース Rev.25
〜 リスクをヘッジしコンプライアンスを堅持するフレームワーク 〜
21CFR Part 820.30 / 75 ISO13485 7.3 / 7.5.6
QSR内部監査員コースご出席要件コースです
( オンサイト及び過去弊社コースを受講された方は要件を満たします )
☆ 開催概要 ☆
ほとんどの日本企業に於ける医療機器開発・設計管理の仕組みは世界標準にほど遠く、これはグローバルに進出しようとする企業のみならず、患者の命を預かる全ての医療機器メーカーにとって大きなリスクを伴います。米国FDAは自らの査察で、医療機器品質不良の根本原因は上流側の設計管理にあると査察の結果から統計的に見抜いており、それはクオリス・イノーバの10年以上にわたるGAP監査でも明らかです。そこで本コースでは、グローバルな開発、設計管理を米国FDA「設計管理ガイダンス」及び欧米で取り入れられている開発フレームワークを元に体系的に解説、また、品質不良を発生させない仕組み(ロバスト設計)をタグチメソッドと設計CAPAを用いて解説します。 2日目はプロセスバリデーションの本質、工程内試験に頼らない均一で高品質な製品を一貫して製造する為、パラメータによる製造方法を実現する工程設計について解説します。 ”規制” だからではなく、設計品質を高めることにより患者さんの命を守り、企業にとっても大きな潜在的利益を得られるという大原則は、エンジニアの方々から多くの共感と支持を頂いてきたクオリス・イノーバの最も人気のある価値あるコースです。 このコースはオンサイトでも実施しております。お問い合わせはこちらから。
日 時: 2021年6月28日(月曜)、29日(火曜) 10:00〜16:30
会 場: 全国町村会館 大ホール(東京・永田町) 永田町駅から徒歩1分( 3番出口 )
講 師: クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象: 医療機器・IVD市場にこれから参入しようとしている企業
医療機器・IVDを海外に輸出しようとしている企業
FDA査察準備、MDAPを受審しようと準備している企業
既にFDA査察を終え、指摘対応に苦慮している企業
受講料: ¥99,000( テキスト・昼食含む )
割 引: ¥85,000( テキスト・朝食含む )
1社2名様以上受講される場合
定 員: 24名
出席者全員に教育記録として利用できる修了書をお渡しします
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡いたします。
2名様以上ご参加の場合、お名前とローマ字読み、メールアドレスを連絡欄にご記入下さい。
持参品: 弊社QSR翻訳本第5版をご持参下さい。お持ちでない方はこちらからお求め頂けます。
ご注意: 本コースは、品質システムの一部を解説するため、システム全体の知識がないと理解できません。本コース受講前に9月開催QSR2日間コースの受講をお勧めします。( QSR2日間コースで
訳本を配布しています )
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1日目(セミナー内容)
A.設計不良はなぜ発生し、再発するのか?
ケーススタディー
直接原因と根本原因は異なる
繰り返される設計品質に起因する製品不良
ISOとQSRの本質的な相違
規制・規格に満足すればOKか?
理解すべき規制・規格・ガイダンス
設計品質の本質的価値
1.Subpart C 設計管理
Waterfall Model と 820.30
ISO13485 vs FDA QSR
FY 483 解析から理解すること
2.820.30 (a) 総則
手順を確立し維持するとは
重要な品質要素、規制要求事項
経営者が提供する設計環境
経営者の責任
プロセスアプローチに基づくリスクヘッジと品質計画
タートルチャートによるリソースとKPIの見定め
リソースとしての要員( 820.25 )
教育と訓練( 訓練の重要性 )
文書体系の基本
3.820.30 (b) 設計及び開発計画
医療機器開発ライフサイクルマネジメント(LCM)
設計品質確立の重要な要素、コンカレントエンジニアリング(CE)
PM、PLの役割
ライフサイクルマネジメント・リスクマネジメント
タグチメソッドの開発方式によるフロントローディング手法
カスタマーニーズを要求仕様に変換するQFD
設計計画書とは
4.820.30 (c) 設計インプット
コミュニケーションエラー
要求仕様書から設計仕様書への変換
曖昧なニーズのスペックへの変換
重要な設計要素トレーサビリティーマトリックス(TMX)
ラベリング、包装設計のインプット
5.820.30 (d) 設計アウトプット
入力のバラツキを押さえたアウトプットの確認
設計アウトプットを定義しアウトプット文書を明確にする
E-BOM の役割
製造仕様書とは
図面発行
DMRとトレーサビリティー
6.820.30 (e) 設計レビュー
問題の早期発見、解決が目的でセレモニーで終わらせない
設計審査で設計のロバストネスを確認
設計審査とテクニカルレビュー
審査者の力量
7.820.30 (f) 設計検証
設計検証と設計バリデーションの相違
インプットからアウトプットまでのトレーサビリティー
エビデンスに基づく設計
設計検証計画に必要な要素
設計検証レポートはデータの信頼性を確保
8.820.30 (g) 設計バリデーション
設計バリデーションの手法
社内、臨床評価の相違と記録
ソフトウェアのバリデーションとVモデル
3種類のバリデーションの相違
9.820.30 (h) 設計移管 → 第2日目(工程設計)
10.820.30 (i ) 設計変更
DHF / DMR / DHR の定義
識別、ファイルの基本
文書体系と設計標準
2日目(セミナー内容)
設計移管と工程設計
1.なぜ工程バリデーションを実施するのか
プロセスバリデーションの目的とその本質
プロセスバリデーションの効果
工程管理、監視の目的と本質
医療機器規制・規格の理解
2.バリデーションすべき工程とは
バリデーションすべき工程の識別
検証工程とバリデーション工程の相違
設備バリデーションと工程バリデーション
バリデーション工程例
3.バリデーションの種類
バリデーションの種類
設備バリデーションと劣化パラメータ
コンピューターシステムバリデーション(CSV)
4.工程設計とバリデーションステップ
VMP:バリデーションマスタープラン
DQ: 設計時適格性評価
IQ: 据付時適格性評価
OQ: 稼働時適格性評価
PQ: 製造時適格性評価
PPQ: プロセス街道性評価
バリデーション文書化
プロセスバリデーションの明確化
自動化工程の明確化
CSVカテゴリーの明確化
ソフトウェアのバリデーションの基本
リスクベースCSV
5.820.30 (h) 設計移管
820.75 プロセスバリデーション
820.72 検査、測定および試験装置
設計移管とプロセスバリデーションの関係
EV、PV、検査測定装置の妥当性とは
ミニスケールとフルスケール
工程設計とPTMX
6.バリデーションの実践
バリデーションマスタープラン(VMP)
バリデーション工程の識別
バリデーション計画
設備リストとバリデーション工程リスト
チームビルディング
DQP/DQR
IQP/IQR
OQP/OQR
PQP/PQR
PPQP/PPQR
7.バリデーションアウトプット
工程能力
再バリデーション設定
設備メンテナンス手順
( 内容が変更される場合がございます )

2020_10 設計管理2日間コース
FDA QSR2日間コース Rev.25
〜 医療機器・IVDに求められる規制要求事項の本質を学ぶ 〜
21CFR Part 820
このコースの売り上げ金の一部は人道支援活動に寄付されます
ISO13485 との比較をより詳細に行っておりシステム構築に最適です
設計管理コース、CAPAコースを受講される方も本コースを受講されることをお勧めします
☆ 開催概要 ☆
* FDA 品質システム規制 QSR 対訳・解説本( 第5版 )付き
グローバルに進出しようとする企業にとって 米国は最も大きなマーケットであり、米国市場での成功がグローバルを制するといっても過言ではありません。しかし、米国に上市するには医療機器規制QSRに基づくFDAの査察は避けられません。( MDSAPを受審される企業も必須 )また、医療機器を国内で販売する際にもFDA QSRのシステムフレームワーク、エビデンスベースの考え方を学び取り入れる事は、製品品質確保につながり、リスクの低減、新しいISO13485にも準拠し、特に受託設計、製造会社 ( ODM、OEM ) では会社のプレゼンスを高め、サプライヤ−としての評価を高めます。 クオリス・イノーバのQSRコースは、多くのFDA査察準備、対応の経験や品質管理の経験から、FDA規制のみならず医療機器開発・製造の本質を鋭く説き、多くの企業の皆様の共感を集め支持されて参りました。米国に進出する企業のみならず、医療機器・IVD を開発しようとしている企業の皆様のマインドが大きく変わる価値あるクオリス・イノーバのメインコースです。
オンサイトでも可能です。お問い合わせはこちらから。
日 時:2021年5月17日(月)、18日(火) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
ODM、OEMとして設計・製造を受託している企業
受講料: ¥99,000( 2日分、テキスト・対訳・解説本(5版)・昼食代含む )
割 引: ¥85,000
1社2名様以上ご参加の場合
特 割: 企業で行うプロジェクトの後、継続研修などでご利用の場合、
特別割引を適用させて頂きますのでメールでご相談下さい。
この場合、正規料金をお支払いの方が優先となりご希望に添えない場合が
ございますので予めご了承ください。
定 員: 20名 5月12日締切
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
開催要件;コロナウィルスの対応について
1.大ホールで開催しお一人様1テーブルのご使用となります
2.換気は自動換気システムが作動し、入口の扉を開けて開催します
3.入口にアルコールを設置、手指消毒と服、靴ウラの消毒をお願いします
4.テーブル、席はホテル側で消毒が行われます。
8.講師、スタッフ、出席者全員マスク着用でお願いいたします
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
第1日目 セミナー内容
1.コンプライアンスとインテグリティー
患者の命と会社の危機管理
データインテグリティー
トップのリーダーシップ
2.行政によるミッションの相違
3.米国法規制の理解
4.医療機器規制の理解
5.なぜISOではFDA査察にパスしないのか
6.QSITアプローチとは
7.規制を正しく理解する要素:定義
820.20 経営者の責任
経営者がリードするTQMが基本
マネジメントレビューの本質、プロセスアプローチ
プロセスを常に改善する
820.22 品質監査
効果的な監査手法と監査報告のあり方
内部監査はプロセスの異常を見極める
内部監査員はなぜ異常を見極められないのか
820.25 要員
教育の重要性:教育の手法と記録、力量表、リソースとは
教育と訓練の相違、アウェアネストレーニングの実施と訓練の重要性
820.30 設計管理( 概要のみ:10月開催 設計監理2日コースで詳細解説 )
ライフサイクル製品開発手法、ウオーターフォールモデル、
フロントローディング手法、ロバスト設計
設計検証と設計バリデーションの相違
今までISO になかった設計移管、工程設計とは
設計CAPAとは
820.40 文書管理
グロバールに通用する文書管理
品質を担保するSOPの作成方法
820.50 購買管理
開発購買とその役割、購買プロセス
3購買(製品、サービス、一般)と購買製品検証
新規サプライヤ評価と継続評価
購買情報とは
820.60 識別
混同を防止する為の識別方法とは
820.65 トレーサビリティー
体内埋込型機器だけではなく、変化点を知り品質を高める
820.80 アクセプタンスアクティビティー
3つのアクセプタンスを理解する
Receiving, In-process, and Finished device acceptance
820.86 アクセプタンス状態
状態表示する意味とは
820.90 不適合品
不適合品の処理プロセス
FDAはなぜNCを見たがるのか
グローバル用語を使用し、CAPAとのリンクを示す
820.120 機器のラベリング
リコールが一番多く査察で注目されるラベリングの出納管理
ラベリングの設計とは
ラベルとラベリングの相違
820.130 機器の梱包
梱包箱を見ればどんな設計を行っていたかが判る
設計依託先はISOで十分か?
820.140 取扱い
混同・劣化・損傷・汚染を発生させないためのリスク管理
820.150 保管
リスクベース保管管理
820.160 流通
ユーザーに届けるまでの製品設計
リスクベース流通管理
820.170 据付け
インスタレーション手順書のV&V
820.180 記録
全ての活動が根拠に基づくエビデンスで保証されるための重要な活動
Objective Evidence を示すための記録管理とは
820.181 機器原簿(DMR)
DMRを定義し識別する
DHF,DMR,DHRの相違
820.184 機器履歴簿(DHR)
製造記録の記載方法
コンプライアンス、製品品質を立証するため重要なDHR
820.186 品質システム記録
DHF,DMR,DHR以外の品質記録とは
820.200 付帯サービス
サービス記録の分析とMR報告,MDRの判定
820.250 統計的手法
TQMを進めていく上で最も重要な要素
品質改善に欠かせない統計的手法
第2日目 セミナー内容
820.70 生産及び工程管理
工程設計と工程V&V
( 概要のみ:10月開催 設計監理2日コースで詳細解説 )
工程のリスクマネジメント(PFMEA)
工程を監視、管理する工程パラメータとは
要員、環境、汚染管理
設備の妥当性とは
保守と検査の相違
副資材の定義と管理
820.72 検査・測定及び試験装置
意図した用途への適合性、測定器の精度管理とは
測定器の精度管理と救済処置
820.75 工程の妥当性確認
QSR,医療機器規制の本質、工程バリデーションとは
( 概要のみ:10月開催 設計監理2日コースで詳細解説 )
TQMが目指すべき試験レスとは
バリデーションの手法( VMP,DQ、IQ、OQ、PQ、PPQ )
設備バリデーションとは
CSV( コンピュータシステムバリデーション )とは
820.198 苦情ファイル
査察のキーポイント、苦情管理の目的を理解する
苦情処理とMDR( Part 803 )、CAR( Part 806 ) の関わり
( 概要のみ:1月開催MDR/CARコースで詳細解説 )
820.100 是正処置及び予防処置( CAPA )
FDA警告書で最も多い指摘CAPAの原則を理解する
品質システムの本質、プロセスアプローチの重要な要素
修正、是正、予防の相違を正確に理解しISOマインドを変える
CAPA7ステップを理解する
CAPAの効果(再発防止)を出す仕組み
MRで監視すべきCAPAプロセス
( 概要のみ:8月開催CAPAコースで詳細解説 )
( 内容が変更になる場合もございますが大きな変更はございません。)
2020_9 QSR Seminar 様子

〜 指摘の最も多いCAPAの理解と問題の予防、再発防止の仕組み 〜
このコースの売上金の一部は人道支援に寄付されます
* コース概要 *
CAPAプロセスを確認するだけで、その会社の品質システムや品質に対する理解度がよくわかります。CAPAプロセスに問題があれば、品質マネジメントシステムの適切性、妥当性はなく、特にマネジメントレビューがまともに運用されているとは到底思われません。品質マネジメントシステムの原則はプロセスアプローチ、PDCAをまわす為、マネジメントレビューのインプットでプロセスを監視し、改善ツールであるCAPAでプロセスを改善します。当たり前のことがシステムとして運用されていないことを理解している規制当局は、CAPAプロセスにフォーカスして確認しようとします。QMSが機能していないことは昨今の品質に関わる事件から明らかで、医療機器・IVDの場合、命に関わる大問題です。本コースでは今までの内容を大きく変更し、プロセスアプローチの手法に重点を置き、事例を元にCAPAの本質を理解して頂きます。 このコースは受講者と同じ目線の御社事例を用いることでより理解が高まるオンサイトセミナーが可能です。お問合せはこちらから。
日 時:2021年4月19日(月曜)10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 ) ( 永田町駅から徒歩1分 )
講 師:クオリス・イノーバ株式会社代表取締役社長 木村 浩実
対 象:医療機器、IVD、医薬品等に限定せず、CAPAプロセスを学びたい企業
受講料: ¥50,000( テキスト代・昼食含む )
割 引: ¥40,000( テキスト代・昼食含む )
1社2名様以上ご参加の場合
特 割:弊社プロジェクトを実施中の場合や、継続研修をご希望の場合、ご負担軽減のためさらに
割引をさせて頂きますのでお問い合わせください。 この場合、正規料金をお支払いのお
客様が優先され、ご出席できない場合がございますのでご了解ください。
再受講:空席がある場合、特別な料金で再受講可能ですのでお問い合わせください。
併せて受講された日もご記入ください。
定 員:20名様 4月14日締め切り
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
開催要件;コロナウィルスの対応について
1.政府の緊急事態宣言が発令された場合キャンセルとなり延期開催はありません
2.大ホールで開催しお一人様1テーブルのご使用となります
3.換気は自動換気システムが作動し入口の扉を開けて開催します
4.入口にアルコールを設置、手指消毒と服、靴ウラの消毒をお願いします
5.テーブル、席はホテル側で消毒が行われます
6.講師、スタッフ、出席者全員マスク着用でお願いいたします
お申込:セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールで連絡いたします。
複数名ご参加の場合、同様の情報をご記入下さい。 割引料金でご案内いたします。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.なぜFDAはCAPAに注目するのか
2.なぜ品質問題は再発するのか
3.規制・規格を正しく理解する
4.プロセスアプローチの基本
プロセス関連図
タートルチャート
5.TQMの仕組みが不可欠
マネジメントレビュー
PDCAをまわす
プロセス改善ツール
6.用語の正しい理解
修正、是正、予防の相違
逸脱、不適合、逸脱許可と特別採用の相違
修正、手直し、修理の相違
7.CAPAプロセスの IN・OUT
CAPAタートルチャート
不適合処理プロセス
苦情処理プロセス
CAPAプロセス
MDRプロセス
リコールプロセス
8.直接原因と根本原因を理解する
直接原因と根本原因の相違
根本原因の改善がプロセスアプローチの本質
9.予防プロセス
品質管理の原則、予防活動の理解
10.エスカレーション
CAPAエスカレーション
CAPAを上手に運用するための仕組み
11.CAPA7ステップ
Step 1 問題を検出し明確にする
Step 2 問題の原因を詳しく調査する
Step 3 根本原因の特定
Step 4 処置の特定と検証または妥当性確認
Step 5 予防または是正する為の処置
Step 6 処置の結果を文書化
Step 7 有効性の確認
12.SCARの実施
13.設計CAPA
14.CAPAマネジメント
マネジメントレビューの運用方法
15.品質問題の事例に学ぶ
Warning Letter Case Study
* 内容(項目)が変更される場合があります

2020_9_CAPA course
FDA MDR&CAR コース Rev.7
〜 指摘の多い有害事象報告・リコール規制の理解とプロセス構築 〜
21CFR : Part 803, 806
3月に延期開催となりました!
このコースの売り上げ金の一部は人道支援活動に寄付されます
☆ 開催概要 ☆
FDA査察レポートの解析からワースト4位(全体の約10%)に急浮上しているMDR(有害事象報告)、日本でも Warning Letter (警告書)を受けているほとんどの企業がMDRの指摘を受けています。昨今のデータ偽装事件を受け、FDAはこれらにフォーカスし厳しく査察を行っています。( 2018FDAレポート)。これはMDR報告の判断に迷ったら報告するという米国の文化とは異なり、日本のメーカーは情報を積極的に発信せず隠すという古い会社の文化が根強くあることが要因の一つですが、そもそも規制を理解せず正しい判断を行っていないことが大きな原因です。医療機器を作っているメーカーは、患者さんの命を守るという社会的な責任があります。会社を守っているようでも、結果的に会社が倒産に追い込まれることもある非常にリスクの高いプロセスであることを理解すべきです。それには規制の正しい理解、規制を守る仕組みと組織が必要です。このセミナーでは、CAPAサブシステム(苦情、不適合、CAPA)の各プロセスとリンクし、日本のメーカーの Warning Letter (警告書)を引き合いに正しいMDRプロセスと関連するCAR(リコール)プロセスを構築することを理解します。今回は薬機法のGVP規制とのリンクも解説します。
このコースはオンサイトでも実施しています。お問い合わせはこちらから。
日 時:2021年3月8日(月曜)13:00 〜 17:00(半日コース)
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:既に医療機器・IVDを米国に輸出している企業
これから医療機器・IVDを米国輸出する準備を行っている企業
既にFDA査察を終え対応に困っている企業
受講料: ¥42,000-
割 引: ¥38,000-( 1社2名様以上受講される場合 )
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.規制を理解する
事例と統計からみる規制違反
理解すべき規制・規格・ガイダンス
eMDR規制
規制が求めるあるべき組織と力量
2.有害事象報告( MDR )
規制の目的、重要性
定義を理解する
MDRとして報告すべき事象とは
5日報告、30日報告とは
FDAへの報告
業態(使用施設、輸入業者、製造業者)毎の報告内容
3.輸入業者との関係
輸入業者のMDRプロセス把握
プロセスの共有と規制が求める契約
eMDR登録
4.MDRプロセス
P820に基づく苦情処理プロセス(グローバル)
P820に基づく不適合処理プロセス
規制に基づくMDRプロセスとは
5.警告書から理解すべき注意点とプロセスへの展開
Warning Letter ( 警告書 )の事例から理解する
警告書から理解すべき注意点とプロセスへの展開
プロセス構築後のレトロスペクティブな是正とは
6.CAR( リコール )プロセス
改修及び回収(CAR)
用語の定義を理解する
報告義務のないCARとは
CARプロセス
ディシジョンツリー
5.警告書から理解すべき注意点とプロセスへの展開
日本の Warning Letter ( 警告書 )の事例から理解する
警告書から理解すべき注意点とプロセスへの展開
*コース内容が一部変更される場合がございますが大きな変更はございません。

2020_1_MDR/CAR
FDA 設計管理2日間コース Rev.24
〜 リスクをヘッジしコンプライアンスを堅持するフレームワーク 〜
21CFR Part 820.30 / 75
QSR内部監査員コースご出席要件コースです
( オンサイト及び過去弊社コースを受講された方は要件を満たします )
☆ 開催概要 ☆
ほとんどの日本企業に於ける医療機器開発・設計管理の仕組みは世界標準にほど遠く、これはグローバルに進出しようとする企業のみならず、患者の命を預かる全ての医療機器メーカーにとって大きなリスクを伴います。米国FDAは自らの査察で、医療機器品質不良の根本原因は上流側の設計管理にあると査察の結果から統計的に見抜いており、それはクオリス・イノーバの10年以上にわたるGAP監査でも明らかです。そこで本コースでは、グローバルな開発、設計管理を米国FDA「設計管理ガイダンス」及び欧米で取り入れられている開発フレームワークを元に体系的に解説、また、品質不良を発生させない仕組み(ロバスト設計)をタグチメソッドと設計CAPAを用いて解説します。 2日目はプロセスバリデーションの本質、工程内試験に頼らない均一で高品質な製品を一貫して製造する為、パラメータによる製造方法を実現する工程設計について解説します。 ”規制” だからではなく、設計品質を高めることにより患者さんの命を守り、企業にとっても大きな潜在的利益を得られるという大原則は、エンジニアの方々から多くの共感と支持を頂いてきたクオリス・イノーバの最も人気のある価値あるコースです。 このコースはオンサイトでも実施しております。お問い合わせはこちらから。
日 時: 2020年10月19日(月曜)、20日(火曜) 10:00〜16:30
会 場: 全国町村会館 大ホール(東京・永田町) 永田町駅から徒歩1分( 3番出口 )
講 師: クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象: 医療機器・IVD市場にこれから参入しようとしている企業
医療機器・IVDを海外に輸出しようとしている企業
FDA査察準備、MDAPを受審しようと準備している企業
既にFDA査察を終え、指摘対応に苦慮している企業
受講料: ¥99,000( テキスト・昼食含む )
割 引: ¥85,000( テキスト・朝食含む )
1社2名様以上受講される場合
定 員: 24名
出席者全員に教育記録として利用できる修了書をお渡しします
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡いたします。
2名様以上ご参加の場合、お名前とローマ字読み、メールアドレスを連絡欄にご記入下さい。
持参品: 弊社QSR翻訳本第4版をご持参下さい。お持ちでない方はこちらからお求め頂けます。
ご注意: 本コースは、品質システムの一部を解説するため、システム全体の知識がないと理解できません。本コース受講前に9月開催QSR2日間コースの受講をお勧めします。( QSR2日間コースで
訳本を配布しています )
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1日目(セミナー内容)
A.設計不良はなぜ発生し、再発するのか?
ケーススタディー
直接原因と根本原因は異なる
繰り返される設計品質に起因する製品不良
ISOとQSRの本質的な相違
規制・規格に満足すればOKか?
理解すべき規制・規格・ガイダンス
設計品質の本質的価値
1.Subpart C 設計管理
Waterfall Model と 820.30
ISO13485 vs FDA QSR
FY 483 解析から理解すること
2.820.30 (a) 総則
手順を確立し維持するとは
重要な品質要素、規制要求事項
経営者が提供する設計環境
経営者の責任
プロセスアプローチに基づくリスクヘッジと品質計画
タートルチャートによるリソースとKPIの見定め
リソースとしての要員( 820.25 )
教育と訓練( 訓練の重要性 )
文書体系の基本
3.820.30 (b) 設計及び開発計画
医療機器開発ライフサイクルマネジメント(LCM)
設計品質確立の重要な要素、コンカレントエンジニアリング(CE)
PM、PLの役割
ライフサイクルマネジメント・リスクマネジメント
タグチメソッドの開発方式によるフロントローディング手法
カスタマーニーズを要求仕様に変換するQFD
設計計画書とは
4.820.30 (c) 設計インプット
コミュニケーションエラー
要求仕様書から設計仕様書への変換
曖昧なニーズのスペックへの変換
重要な設計要素トレーサビリティーマトリックス(TMX)
ラベリング、包装設計のインプット
5.820.30 (d) 設計アウトプット
入力のバラツキを押さえたアウトプットの確認
設計アウトプットを定義しアウトプット文書を明確にする
E-BOM の役割
製造仕様書とは
図面発行
DMRとトレーサビリティー
6.820.30 (e) 設計レビュー
問題の早期発見、解決が目的でセレモニーで終わらせない
設計審査で設計のロバストネスを確認
設計審査とテクニカルレビュー
審査者の力量
7.820.30 (f) 設計検証
設計検証と設計バリデーションの相違
インプットからアウトプットまでのトレーサビリティー
エビデンスに基づく設計
設計検証計画に必要な要素
設計検証レポートはデータの信頼性を確保
8.820.30 (g) 設計バリデーション
設計バリデーションの手法
社内、臨床評価の相違と記録
ソフトウェアのバリデーションとVモデル
3種類のバリデーションの相違
9.820.30 (h) 設計移管 → 第2日目(工程設計)
10.820.30 (i ) 設計変更
DHF / DMR / DHR の定義
識別、ファイルの基本
文書体系と設計標準
2日目(セミナー内容)
設計移管と工程設計
1.なぜ工程バリデーションを実施するのか
プロセスバリデーションの目的とその本質
プロセスバリデーションの効果
工程管理、監視の目的と本質
医療機器規制・規格の理解
2.バリデーションすべき工程とは
バリデーションすべき工程の識別
検証工程とバリデーション工程の相違
設備バリデーションと工程バリデーション
バリデーション工程例
3.バリデーションの種類
バリデーションの種類
設備バリデーションと劣化パラメータ
コンピューターシステムバリデーション(CSV)
4.工程設計とバリデーションステップ
VMP:バリデーションマスタープラン
DQ: 設計時適格性評価
IQ: 据付時適格性評価
OQ: 稼働時適格性評価
PQ: 製造時適格性評価
PPQ: プロセス街道性評価
バリデーション文書化
プロセスバリデーションの明確化
自動化工程の明確化
CSVカテゴリーの明確化
ソフトウェアのバリデーションの基本
リスクベースCSV
5.820.30 (h) 設計移管
820.75 プロセスバリデーション
820.72 検査、測定および試験装置
設計移管とプロセスバリデーションの関係
EV、PV、検査測定装置の妥当性とは
ミニスケールとフルスケール
工程設計とPTMX
6.バリデーションの実践
バリデーションマスタープラン(VMP)
バリデーション工程の識別
バリデーション計画
設備リストとバリデーション工程リスト
チームビルディング
DQP/DQR
IQP/IQR
OQP/OQR
PQP/PQR
PPQP/PPQR
7.バリデーションアウトプット
工程能力
再バリデーション設定
設備メンテナンス手順
( 内容が変更される場合がございます )
 2019_10 設計管理2日間コース
2019_10 設計管理2日間コース
QMS 購買管理コース
〜 SCM でQCDに貢献する購買プロセスの本質 〜
21CFR Part 820・ISO13485・ISO9001
このコースの売り上げ金の一部は人道支援活動に寄付されます
弊社コロナウィルス対策はこちらからご確認下さい
☆ 開催概要 ☆
購買プロセスは言われた通りにモノを買っていればよいというものではなく、サプライチェーン全体を俯瞰し、製品企画時の上流から積極的にコンカレントで参画、最も効率よいQCDを提案することこそ会社に貢献するプロセスとして価値があります。その為には、戦略的な組織体制とフレームを構築する必要があり、開発購買や情報購買と言ったフレームワークは早期の製品ローンチを実現します。また、アッセンブルラーが多くなった昨今、行政当局も購買プロセスに多くの査察時間を費やすようになりました。自社と同じシステムで管理する必要性が高まり、フレームと管理能力が要求されます。FDA QSRのみならず、ISOの基本9001の要求も加味しながら理解を深めます。
オンサイトでも可能です。お問い合わせはこちらから。
日 時:2020年11月5日(木曜) 13:00 〜 16:30 半日コースです
会 場:全国町村会館(東京・永田町) 永田町駅から徒歩1分( 3番出口 )
講 師:クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
ODM、OEMとして設計・製造を受託している企業
受講料: ¥42.000-( 午後半日、テキスト代含む )
割 引: ¥38.000-( 1社2名様以上ご参加の場合 )
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
A.FDA規制、査察の最新情報と傾向分析
1.適用規制・規格
FDA QSR,ISO13485,9001の要求とは
プロセスアプローチでインとアウトを理解する
2.購買管理の目的
規格が示す購買管理の目的
製品、サービス購買とは
4つの購買範囲とは
3.用語の定義
製品、サービスとは
プロバイダー、サプライヤー、コントラクターとは
4.購買管理
GHTF( IMDRF )ガイダンスのフロー
5.購買方針
購買方針がなければ購買プロセスは成り立たない
6.戦略・開発購買
サプライチェーン全体を俯瞰した戦略
開発スピードを上げ原価削減、購買活動のスピードを上げある
ベストプラクティスの収集と技術の取り込み
7.購買計画
製品企画段階から参画
供給する製品、サービスの特定
技術及びプロセス情報の特定
供給者候補の特定
リスク、管理方法の特定
8.供給者候補選定
供給者ビジネス、運用能力評価
供給者候補選定
9.供給者選定
評価選定基準の策定
供給者候補の能力評価
供給者の承認
10.管理方法の取り決め
購買情報とは
管理方法
11.変更時対応
変更管理規定
4Mと変化点管理
12.納期、測定、監視
製品、サービスの検証
不適合の手順
供給者の監視
製品の受領、出荷場、冷蔵室、NCエリア
カーゴ内温度バリデーション
13.Feedback, Communication
CR
SCAR
供給者評価シート
14.Supplier Audit
リスクベース監査
5つの監査、評価エリア
情報購買、戦略上重要な監査チーム
( 内容が変更になる場合もございますが大きな変更はございません。)
2019_07_PC
FDA QSR2日間コース Rev.24
〜 医療機器・IVDに求められる規制要求事項の本質を学ぶ 〜
21CFR Part 820
このコースの売り上げ金の一部は人道支援活動に寄付されます
ISO13485 との比較をより詳細に行いシステム構築に最適です
設計管理コース、CAPAコースを受講される方も本コースを受講されることをお勧めします
本コースは本年は今回の1回開催のみとなります
☆ 開催概要 ☆
* FDA 品質システム規制 QSR 対訳・解説本( 第5版 )付き
グローバルに進出しようとする企業にとって 米国は最も大きなマーケットであり、米国市場での成功がグローバルを制するといっても過言ではありません。しかし、米国に上市するには医療機器規制QSRに基づくFDAの査察は避けられません。( MDSAPを受審される企業も必須 )また、医療機器を国内で販売する際にもFDA QSRのシステムフレームワーク、エビデンスベースの考え方を学び取り入れる事は、製品品質確保につながり、リスクの低減、新しいISO13485にも準拠し、特に受託設計、製造会社 ( ODM、OEM ) では会社のプレゼンスを高め、サプライヤ−としての評価を高めます。 クオリス・イノーバのQSRコースは、多くのFDA査察準備、対応の経験や品質管理の経験から、FDA規制のみならず医療機器開発・製造の本質を鋭く説き、多くの企業の皆様の共感を集め支持されて参りました。米国に進出する企業のみならず、医療機器・IVD を開発しようとしている企業の皆様のマインドが大きく変わる価値あるクオリス・イノーバのメインコースです。
オンサイトでも可能です。お問い合わせはこちらから。
日 時:2020年9月14日(月)、15日(火) 10:00 〜 16:30
6月開催から9月に変更しました( 5月27日アナウンス )
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
ODM、OEMとして設計・製造を受託している企業
受講料: ¥99,000( 2日分、テキスト・対訳・解説本(5版)・昼食代含む )
割 引: ¥85,000
1社2名様以上ご参加の場合
特 割: 企業で行うプロジェクトの後、継続研修などでご利用の場合、
特別割引を適用させて頂きますのでメールでご相談下さい。
この場合、正規料金をお支払いの方が優先となりご希望に添えない場合が
ございますので予めご了承ください。
定 員: 20名 9月7日締切
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
開催要件;コロナウィルスの対応について
1.政府の緊急事態宣言が発令された場合キャンセルとなり延期開催はありません
2.キャンセルの可能性が否定できないため事前のご入金はありません
3.大ホールで開催しお一人様1テーブルのご使用となります
4.換気は自動換気システムが作動し、入口の扉を開けて開催します
5.プロジェクターを3台用意し、広い会場で不便の内容配慮します
6.入口にアルコールを設置、手指消毒と服、靴ウラの消毒をお願いします
7.テーブル、席はホテル側で消毒が行われます
8.講師、スタッフ、出席者全員マスク着用でお願いいたします
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
第1日目 セミナー内容
1.コンプライアンスとインテグリティー
患者の命と会社の危機管理
データインテグリティー
トップのリーダーシップ
2.行政によるミッションの相違
3.米国法規制の理解
4.医療機器医規制の理解
5.なぜISOではFDA査察にパスしないのか
6.QSITアプローチとは
7.規制を正しく理解する要素:定義
820.20 経営者の責任
経営者がリードするTQMが基本
マネジメントレビューの本質、プロセスアプローチ
プロセスを常に改善する
820.22 品質監査
効果的な監査手法と監査報告のあり方
内部監査はプロセスの異常を見極める
内部監査員はなぜ異常を見極められないのか
820.25 要員
教育の重要性:教育の手法と記録、力量表、リソースとは
教育と訓練の相違、アウェアネストレーニングの実施と訓練の重要性
820.30 設計管理( 概要のみ:10月開催 設計監理2日コースで詳細解説 )
ライフサイクル製品開発手法、ウオーターフォールモデル、
フロントローディング手法、ロバスト設計
設計検証と設計バリデーションの相違
今までISO になかった設計移管、工程設計とは
設計CAPAとは
820.40 文書管理
グロバールに通用する文書管理
品質を担保するSOPの作成方法
820.50 購買管理
開発購買とその役割、購買プロセス
3つの購買(製品、サービス、一般)と購買製品検証
新規サプライヤ評価と継続評価
購買情報とは
820.60 識別
混同を防止する為の識別方法とは
820.65 トレーサビリティー
体内埋込型機器だけではなく、変化点を知り品質を高める
820.80 アクセプタンスアクティビティー
3つのアクセプタンスを理解する
Receiving, In-process, and Finished device acceptance
820.86 アクセプタンス状態
状態表示する意味とは
820.90 不適合品
不適合品の処理プロセス
FDAはなぜNCを見たがるのか
グローバル用語を使用し、CAPAとのリンクを示す
820.120 機器のラベリング
リコールが一番多く査察で注目されるラベリングの出納管理
ラベリングの設計とは
ラベルとラベリングの相違
820.130 機器の梱包
梱包箱を見ればどんな設計を行っていたかが判る
設計依託先はISOで十分か?
820.140 取扱い
混同・劣化・損傷・汚染を発生させないためのリスク管理
820.150 保管
リスクベース保管管理
820.160 流通
ユーザーに届けるまでの製品設計
リスクベース流通管理
820.170 据付け
インスタレーション手順書のV&V
820.180 記録
全ての活動が根拠に基づくエビデンスで保証されるための重要な活動
Objective Evidence を示すための記録管理とは
820.181 機器原簿(DMR)
DMRを定義し識別する
DHF,DMR,DHRの相違
820.184 機器履歴簿(DHR)
製造記録の記載方法
コンプライアンス、製品品質を立証するため重要なDHR
820.186 品質システム記録
DHF,DMR,DHR以外の品質記録とは
820.200 付帯サービス
サービス記録の分析とMR報告,MDRの判定
820.250 統計的手法
TQMを進めていく上で最も重要な要素
品質改善に欠かせない統計的手法
第2日目 セミナー内容
820.70 生産及び工程管理
工程設計と工程V&V
( 概要のみ:10月開催 設計監理2日コースで詳細解説 )
工程のリスクマネジメント(PFMEA)
工程を監視、管理する工程パラメータとは
要員、環境、汚染管理
設備の妥当性とは
保守と検査の相違
副資材の定義と管理
820.72 検査・測定及び試験装置
意図した用途への適合性、測定器の精度管理とは
測定器の精度管理と救済処置
820.75 工程の妥当性確認
QSR,医療機器規制の本質、工程バリデーションとは
( 概要のみ:10月開催 設計監理2日コースで詳細解説 )
TQMが目指すべき試験レスとは
バリデーションの手法( VMP,DQ、IQ、OQ、PQ、PPQ )
設備バリデーションとは
CSV( コンピュータシステムバリデーション )とは
820.198 苦情ファイル
査察のキーポイント、苦情管理の目的を理解する
苦情処理とMDR( Part 803 )、CAR( Part 806 ) の関わり
( 概要のみ:1月開催MDR/CARコースで詳細解説 )
820.100 是正処置及び予防処置( CAPA )
FDA警告書で最も多い指摘CAPAの原則を理解する
品質システムの本質、プロセスアプローチの重要な要素
修正、是正、予防の相違を正確に理解しISOマインドを変える
CAPA7ステップを理解する
CAPAの効果(再発防止)を出す仕組み
MRで監視すべきCAPAプロセス
( 概要のみ:8月開催CAPAコースで詳細解説 )
( 内容が変更になる場合もございますが大きな変更はございません。)
2019_9 QSR Seminar 様子

CAPAコース Rev.19
〜 指摘の最も多いCAPAの理解と問題の予防、再発防止の仕組み 〜
このコースの売上金の一部は人道支援に寄付されます
本コースは6月8日に開催予定でしたが、
コロナウィルスの影響で
8月31日延期開催することになりました
* コース概要 *
CAPAプロセスを確認するだけで、その会社の品質システムや品質に対する理解度がよくわかります。CAPAプロセスに問題があれば、品質マネジメントシステムの適切性、妥当性はなく、特にマネジメントレビューがまともに運用されているとは到底思われません。品質マネジメントシステムの原則はプロセスアプローチ、PDCAをまわす為、マネジメントレビューのインプットでプロセスを監視し、改善ツールであるCAPAでプロセスを改善します。当たり前のことがシステムとして運用されていないことを理解している規制当局は、CAPAプロセスにフォーカスして確認しようとします。QMSが機能していないことは昨今の品質に関わる事件から明らかで、医療機器・IVDの場合、命に関わる大問題です。本コースでは今までの内容を大きく変更し、プロセスアプローチの手法に重点を置き、事例を元にCAPAの本質を理解して頂きます。 このコースは受講者と同じ目線の御社事例を用いることでより理解が高まるオンサイトセミナーが可能です。お問合せはこちらから。
日 時:2020年8月31日(月曜)10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 ) ( 永田町駅から徒歩1分 )
講 師:クオリス・イノーバ株式会社代表取締役社長 木村 浩実
対 象:医療機器、IVD、医薬品等に限定せず、CAPAプロセスを学びたい企業
受講料: ¥50,000( テキスト代・昼食含む )
割 引: ¥40,000( テキスト代・昼食含む )
1社2名様以上ご参加の場合
特 割:弊社プロジェクトを実施中の場合や、継続研修をご希望の場合、ご負担軽減のためさらに
割引をさせて頂きますのでお問い合わせください。 この場合、正規料金をお支払いのお
客様が優先され、ご出席できない場合がございますのでご了解ください。
再受講:空席がある場合、特別な料金で再受講可能ですのでお問い合わせください。
併せて受講された日もご記入ください。
定 員:20名様 8月17日締め切り
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
開催要件;コロナウィルスの対応について
1.政府の緊急事態宣言が発令された場合キャンセルとなり延期開催はありません
2.キャンセルの可能性が否定できないため事前のご入金はありません
3.大ホールで開催しお一人様1テーブルのご使用となります
4.換気は自動換気システムが作動し、入口の扉を開けて開催します
5.プロジェクターを3台用意し、広い会場で不便の内容配慮します
6.入口にアルコールを設置、手指消毒と服、靴ウラの消毒をお願いします
7.テーブル、席はホテル側で消毒が行われます
8.講師、スタッフ、出席者全員マスク着用でお願いいたします
お申込:セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールで連絡いたします。
複数名ご参加の場合、同様の情報をご記入下さい。 割引料金でご案内いたします。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.なぜFDAはCAPAに注目するのか
2.なぜ品質問題は再発するのか
3.規制・規格を正しく理解する
4.プロセスアプローチの基本
プロセス関連図
タートルチャート
5.TQMの仕組みが不可欠
マネジメントレビュー
PDCAをまわす
プロセス改善ツール
6.用語の正しい理解
修正、是正、予防の相違
逸脱、不適合、逸脱許可と特別採用の相違
修正、手直し、修理の相違
7.CAPAプロセスの IN・OUT
CAPAタートルチャート
不適合処理プロセス
苦情処理プロセス
CAPAプロセス
MDRプロセス
リコールプロセス
8.直接原因と根本原因を理解する
直接原因と根本原因の相違
根本原因の改善がプロセスアプローチの本質
9.予防プロセス
品質管理の原則、予防活動の理解
10.エスカレーション
CAPAエスカレーション
CAPAを上手に運用するための仕組み
11.CAPA7ステップ
Step 1 問題を検出し明確にする
Step 2 問題の原因を詳しく調査する
Step 3 根本原因の特定
Step 4 処置の特定と検証または妥当性確認
Step 5 予防または是正する為の処置
Step 6 処置の結果を文書化
Step 7 有効性の確認
12.CARの実施
13.設計CAPA
14.CAPAマネジメント
マネジメントレビューの運用方法
15.品質問題の事例に学ぶ
Warning Letter Case Study
* 内容(項目)が変更される場合があります

2019_3_CAPA course
FDA MDR&CAR コース Rev.6
〜 指摘の多い有害事象報告・リコール規制の理解とプロセス構築 〜
21CFR : Part 803, 806
このコースの売り上げ金の一部は人道支援活動に寄付されます
☆ 開催概要 ☆
FDA査察レポートの解析からワースト4位(全体の約10%)に急浮上しているMDR(有害事象報告)、日本でも Warning Letter (警告書)を受けているほとんどの企業がMDRの指摘を受けています。昨今のデータ偽装事件を受け、FDAはこれらにフォーカスし厳しく査察を行っています。( 2018FDAレポート)。これはMDR報告の判断に迷ったら報告するという米国の文化とは異なり、日本のメーカーは情報を積極的に発信せず隠すという古い会社の文化が根強くあることが要因の一つですが、そもそも規制を理解せず正しい判断を行っていないことが大きな原因です。医療機器を作っているメーカーは、患者さんの命を守るという社会的な責任があります。会社を守っているようでも、結果的に会社が倒産に追い込まれることもある非常にリスクの高いプロセスであることを理解すべきです。それには規制の正しい理解、規制を守る仕組みと組織が必要です。このセミナーでは、CAPAサブシステム(苦情、不適合、CAPA)の各プロセスとリンクし、日本のメーカーの Warning Letter (警告書)を引き合いに正しいMDRプロセスと関連するCAR(リコール)プロセスを構築することを理解します。
このコースはオンサイトでも実施しています。お問い合わせはこちらから。
日 時:2020年1月27日(月曜)13:00 〜 16:30(半日コース)
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:既に医療機器・IVDを米国に輸出している企業
これから医療機器・IVDを米国輸出する準備を行っている企業
既にFDA査察を終え対応に困っている企業
受講料: ¥42,000-
割 引: ¥38,000-( 1社2名様以上受講される場合 )
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.規制を理解する
事例と統計からみる規制違反
理解すべき規制・規格・ガイダンス
eMDR規制
規制が求めるあるべき組織と力量
2.有害事象報告( MDR )
規制の目的、重要性
MDRとして報告すべき事象とは
5日報告、30日報告とは
業態(使用施設、輸入業者、製造業者)毎の報告内容
3.輸入業者との関係
輸入業者のMDRプロセス把握
プロセスの共有と規制が求める契約
eMDR登録
4.MDRプロセス
P820に基づく苦情処理プロセス(グローバル)
P820に基づく不適合処理プロセス
規制に基づくMDRプロセスとは
5.警告書から理解すべき注意点とプロセスへの展開
Warning Letter ( 警告書 )の事例から理解する
警告書から理解すべき注意点とプロセスへの展開
プロセス構築後のレトロスペクティブな是正とは
6.CAR( リコール )プロセス
改修及び回収(CAR)
用語の定義を理解する
報告義務のないCARとは
CARプロセス
ディシジョンツリー
5.警告書から理解すべき注意点とプロセスへの展開
日本の Warning Letter ( 警告書 )の事例から理解する
警告書から理解すべき注意点とプロセスへの展開
*コース内容が一部変更される場合がございますが大きな変更はございません。

2019 MDR/CAR
FDA 設計管理2日間コース Rev.23
〜 リスクをヘッジしコンプライアンスを堅持するフレームワーク 〜
21CFR Part 820.30 / 75
12月開催、QSR内部監査員コースご出席要件コースです
( オンサイト及び過去弊社コースを受講された方は要件を満たします )
定員オーバーのため募集を終了致しました
次回は来年5月以降となります(募集は3月から)
☆ 開催概要 ☆
ほとんどの日本企業に於ける医療機器開発・設計管理の仕組みは世界標準にほど遠く、これはグローバルに進出しようとする企業のみならず、患者の命を預かる全ての医療機器メーカーにとって大きなリスクを伴います。米国FDAは自らの査察で、医療機器品質不良の根本原因は上流側の設計管理にあると査察の結果から統計的に見抜いており、それはクオリス・イノーバの10年以上にわたるGAP監査でも明らかです。そこで本コースでは、グローバルな開発、設計管理を米国FDA「設計管理ガイダンス」及び欧米で取り入れられている開発フレームワークを元に体系的に解説、また、品質不良を発生させない仕組み(ロバスト設計)をタグチメソッドと設計CAPAを用いて解説します。 2日目はプロセスバリデーションの本質、工程内試験に頼らない均一で高品質な製品を一貫して製造する為、パラメータによる製造方法を実現する工程設計について解説します。 ”規制” だから、ではなく、設計品質を高めることにより患者さんの命を守り、企業にとっても大きな潜在的利益を得られるという大原則は、エンジニアの方々から多くの共感と支持を頂いてきたクオリス・イノーバの最も人気のある価値あるコースです。 このコースはオンサイトでも実施しております。お問い合わせはこちらから。
日 時: 2019年10月24日(木曜)、25日(金曜) 10:00〜16:30
会 場: 全国町村会館(東京・永田町) 永田町駅から徒歩1分( 3番出口 )
講 師: クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象: 医療機器・IVD市場にこれから参入しようとしている企業
医療機器・IVDを海外に輸出しようとしている企業
FDA査察準備、MDAPを受審しようと準備している企業
既にFDA査察を終え、指摘対応に苦慮している企業
受講料: ¥99,000( テキスト・昼食含む )
割 引: ¥85,000( テキスト・朝食含む )
1社2名様以上受講される場合
定 員: 24名
出席者全員に教育記録として利用できる修了書をお渡しします
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡いたします。
2名様以上ご参加の場合、お名前とローマ字読み、メールアドレスを連絡欄にご記入下さい。
持参品: 弊社QSR翻訳本第三版をご持参下さい。お持ちでない方はこちらからお求め頂けます。
ご注意: 本コースは、品質システムの一部を解説するため、システム全体の知識がないと理解できません。本コース受講前に9月開催QSR2日間コースの受講をお勧めします。( QSR2日間コースで
訳本を配布しています )
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1日目(セミナー内容)
A.設計不良はなぜ発生し、再発するのか?
ケーススタディー
直接原因と根本原因は異なる
繰り返される設計品質に起因する製品不良
ISOとQSRの本質的な相違
規制・規格に満足すればOKか?
理解すべき規制・規格・ガイダンス
設計品質の本質的価値
1.Subpart C 設計管理
Waterfall Model と 820.30
ISO13485 vs FDA QSR
FY 483 解析から理解すること
2.820.30 (a) 総則
手順を確立し維持するとは
重要な品質要素、規制要求事項
経営者が提供する設計環境
経営者の責任
プロセスアプローチに基づくリスクヘッジと品質計画
タートルチャートによるリソースとKPIの見定め
リソースとしての要員( 820.25 )
教育と訓練( 訓練の重要性 )
文書体系の基本
3.820.30 (b) 設計及び開発計画
医療機器開発ライフサイクルマネジメント(LCM)
設計品質確立の重要な要素、コンカレントエンジニアリング(CE)
PM、PLの役割
ライフサイクルマネジメント・リスクマネジメント
タグチメソッドの開発方式によるフロントローディング手法
カスタマーニーズを要求仕様に変換するQFD
設計計画書とは
4.820.30 (c) 設計インプット
コミュニケーションエラー
要求仕様書から設計仕様書への変換
曖昧なニーズのスペックへの変換
重要な設計要素トレーサビリティーマトリックス(TMX)
ラベリング、包装設計のインプット
5.820.30 (d) 設計アウトプット
入力のバラツキを押さえたアウトプットの確認
設計アウトプットを定義しアウトプット文書を明確にする
E-BOM の役割
製造仕様書とは
図面発行
DMRとトレーサビリティー
6.820.30 (e) 設計レビュー
問題の早期発見、解決が目的でセレモニーで終わらせない
設計審査で設計のロバストネスを確認
設計審査とテクニカルレビュー
審査者の力量
7.820.30 (f) 設計検証
設計検証と設計バリデーションの相違
インプットからアウトプットまでのトレーサビリティー
エビデンスに基づく設計
設計検証計画に必要な要素
設計検証レポートはデータの信頼性を確保
8.820.30 (g) 設計バリデーション
設計バリデーションの手法
社内、臨床評価の相違と記録
ソフトウェアのバリデーションとVモデル
3種類のバリデーションの相違
9.820.30 (h) 設計移管 → 第2日目(工程設計)
10.820.30 (i ) 設計変更
DHF / DMR / DHR の定義
識別、ファイルの基本
文書体系と設計標準
2日目(セミナー内容)
設計移管と工程設計
1.なぜ工程バリデーションを実施するのか
プロセスバリデーションの目的とその本質
プロセスバリデーションの効果
工程管理、監視の目的と本質
医療機器規制・規格の理解
2.バリデーションすべき工程とは
バリデーションすべき工程の識別
検証工程とバリデーション工程の相違
設備バリデーションと工程バリデーション
バリデーション工程例
3.バリデーションの種類
バリデーションの種類
設備バリデーションと劣化パラメータ
コンピューターシステムバリデーション(CSV)
4.工程設計とバリデーションステップ
VMP:バリデーションマスタープラン
DQ: 設計時適格性評価
IQ: 据付時適格性評価
OQ: 稼働時適格性評価
PQ: 製造時適格性評価
PPQ: プロセス街道性評価
バリデーション文書化
プロセスバリデーションの明確化
自動化工程の明確化
CSVカテゴリーの明確化
ソフトウェアのバリデーションの基本
リスクベースCSV
5.820.30 (h) 設計移管
820.75 プロセスバリデーション
820.72 検査、測定および試験装置
設計移管とプロセスバリデーションの関係
EV、PV、検査測定装置の妥当性とは
ミニスケールとフルスケール
工程設計とPTMX
6.バリデーションの実践
バリデーションマスタープラン(VMP)
バリデーション工程の識別
バリデーション計画
設備リストとバリデーション工程リスト
チームビルディング
DQP/DQR
IQP/IQR
OQP/OQR
PQP/PQR
PPQP/PPQR
7.バリデーションアウトプット
工程能力
再バリデーション設定
設備メンテナンス手順
( 内容が変更される場合がございます )
 2019_6 設計管理2日間コース
2019_6 設計管理2日間コース
FDA QSR2日間コース Rev.23
〜 医療機器・IVDに求められる規制要求事項の本質を学ぶ 〜
21CFR Part 820
このコースの売り上げ金の一部は人道支援活動に寄付されます
ISO13485 との比較をより詳細に行いシステム構築に最適です
12月開催 FDA 内部監査員コースの出席される場合の要件コースです
設計管理コース、CAPAコースを受講される方も本コースを受講されることをお勧めします
定員オーバーのため募集を締め切らせて頂きました
次回開催は来年4月以降となります
☆ 開催概要 ☆
* FDA 品質システム規制 QSR 対訳本( 第3版 )付き
グローバルに進出しようとする企業にとって 米国は最も大きなマーケットであり、米国市場での成功がグローバルを制するといっても過言ではありません。しかし、米国に上市するには医療機器規制QSRに基づくFDAの査察は避けられません。( MDSAPを受審される企業も必須 )また、医療機器を国内で販売する際にもFDA QSRのシステムフレームワーク、エビデンスベースの考え方を学び取り入れる事は、製品品質確保につながり、リスクの低減、新しいISO13485にも準拠し、特に受託設計、製造会社 ( ODM、OEM ) では会社のプレゼンスを高め、サプライヤ−としての評価を高めます。 クオリス・イノーバのQSRコースは、多くのFDA査察準備、対応の経験や品質管理の経験から、FDA規制のみならず医療機器開発・製造の本質を鋭く説き、多くの企業の皆様の共感を集め支持されて参りました。米国に進出する企業のみならず、医療機器・IVD を開発しようとしている企業の皆様のマインドが大きく変わる価値あるクオリス・イノーバのメインコースです。
オンサイトでも可能です。お問い合わせはこちらから。
日 時:2019年9月17日(火)、18日(水) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
ODM、OEMとして設計・製造を受託している企業
受講料: ¥99,000( 2日分、テキスト・対訳本(3版)・昼食代含む )
割 引: ¥85,000
1社2名様以上ご参加の場合
特 割: 企業で行うプロジェクトの後、継続研修などでご利用の場合、
特別割引を適用させて頂きますのでメールでご相談下さい。
この場合、正規料金をお支払いの方が優先となりご希望に添えない場合が
ございますので予めご了承ください。
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
第1日目 セミナー内容
1.コンプライアンスとインテグリティー
患者の命と会社の危機管理
データインテグリティー
トップのリーダーシップ
2.行政によるミッションの相違
3.米国法規制の理解
4.医療機器医規制の理解
5.なぜISOではFDA査察にパスしないのか
6.QSITアプローチとは
7.規制を正しく理解する要素:定義
820.20 経営者の責任
経営者がリードするTQMが基本
マネジメントレビューの本質、プロセスアプローチ
プロセスを常に改善する
820.22 品質監査
効果的な監査手法と監査報告のあり方
内部監査はプロセスの異常を見極める
内部監査員はなぜ異常を見極められないのか
820.25 要員
教育の重要性:教育の手法と記録、力量表、リソースとは
教育と訓練の相違、アウェアネストレーニングの実施と訓練の重要性
820.30 設計管理( 概要のみ:10月開催 設計監理2日コースで詳細解説 )
ライフサイクル製品開発手法、ウオーターフォールモデル、
フロントローディング手法、ロバスト設計
設計検証と設計バリデーションの相違
今までISO になかった設計移管、工程設計とは
設計CAPAとは
820.40 文書管理
グロバールに通用する文書管理
品質を担保するSOPの作成方法
820.50 購買管理
開発購買とその役割、購買プロセス
3つの購買(製品、サービス、一般)と購買製品検証
新規サプライヤ評価と継続評価
購買情報とは
820.60 識別
混同を防止する為の識別方法とは
820.65 トレーサビリティー
体内埋込型機器だけではなく、変化点を知り品質を高める
820.80 アクセプタンスアクティビティー
3つのアクセプタンスを理解する
Receiving, In-process, and Finished device acceptance
820.86 アクセプタンス状態
状態表示する意味とは
820.90 不適合品
不適合品の処理プロセス
FDAはなぜNCを見たがるのか
グローバル用語を使用し、CAPAとのリンクを示す
820.120 機器のラベリング
リコールが一番多く査察で注目されるラベリングの出納管理
ラベリングの設計とは
ラベルとラベリングの相違
820.130 機器の梱包
梱包箱を見ればどんな設計を行っていたかが判る
設計依託先はISOで十分か?
820.140 取扱い
混同・劣化・損傷・汚染を発生させないためのリスク管理
820.150 保管
リスクベース保管管理
820.160 流通
ユーザーに届けるまでの製品設計
リスクベース流通管理
820.170 据付け
インスタレーション手順書のV&V
820.180 記録
全ての活動が根拠に基づくエビデンスで保証されるための重要な活動
Objective Evidence を示すための記録管理とは
820.181 機器原簿(DMR)
DMRを定義し識別する
DHF,DMR,DHRの相違
820.184 機器履歴簿(DHR)
製造記録の記載方法
コンプライアンス、製品品質を立証するため重要なDHR
820.186 品質システム記録
DHF,DMR,DHR以外の品質記録とは
820.200 付帯サービス
サービス記録の分析とMR報告,MDRの判定
820.250 統計的手法
TQMを進めていく上で最も重要な要素
品質改善に欠かせない統計的手法
第2日目 セミナー内容
820.70 生産及び工程管理
工程設計と工程V&V
( 概要のみ:10月開催 設計監理2日コースで詳細解説 )
工程のリスクマネジメント(PFMEA)
工程を監視、管理する工程パラメータとは
要員、環境、汚染管理
設備の妥当性とは
保守と検査の相違
副資材の定義と管理
820.72 検査・測定及び試験装置
意図した用途への適合性、測定器の精度管理とは
測定器の精度管理と救済処置
820.75 工程の妥当性確認
QSR,医療機器規制の本質、工程バリデーションとは
( 概要のみ:10月開催 設計監理2日コースで詳細解説 )
TQMが目指すべき試験レスとは
バリデーションの手法( VMP,DQ、IQ、OQ、PQ、PPQ )
設備バリデーションとは
CSV( コンピュータシステムバリデーション )とは
820.198 苦情ファイル
査察のキーポイント、苦情管理の目的を理解する
苦情処理とMDR( Part 803 )、CAR( Part 806 ) の関わり
( 概要のみ:1月開催MDR/CARコースで詳細解説 )
820.100 是正処置及び予防処置( CAPA )
FDA警告書で最も多い指摘CAPAの原則を理解する
品質システムの本質、プロセスアプローチの重要な要素
修正、是正、予防の相違を正確に理解しISOマインドを変える
CAPA7ステップを理解する
CAPAの効果(再発防止)を出す仕組み
MRで監視すべきCAPAプロセス
( 概要のみ:3月開催CAPAコースで詳細解説 )
( 内容が変更になる場合もございますが大きな変更はございません。)
2019_5 QSR Seminar 様子

付帯サービスコース
〜 医療機器グローバルサービス設計とデータ運用 〜
21CFR Part 820.200 / ISO13485 7.5.4
このコースの売上金の一部は人道活動支援に寄付されます
午後開催『 QMS購買管理コース 』に続けて出席なさる方には昼食をご用意
定員オーバーのため募集を締め切らせて頂きました
☆ 開催概要 ☆
ほとんどの日本企業に於ける医療機器付帯サービス活動は、コストであって製品としてビジネスが成立していません。また、プロセスとしてコンカレント設計がない企業ではサービス活動は製品設計のインプットになく、付帯サービスで無駄なコストや製品価値の低下を招き、グローバルにもサービス品質で競争に負けています。さらに、サービスデータは開発の上流にフィードバックすべきデータの宝庫で、改善の積み重ねが品質を高め、結果として患者さんの命を守ります。これらは規制、規格でも求められています。
オンサイトでも可能です。お問い合わせはこちらから。
日 時: 2019年7月8日(月曜)9:45 〜 11:45 午前半日コースです
会 場: 全国町村会館(東京・永田町) 永田町駅から徒歩1分( 3番出口 )
講 師: クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象: 医療機器・IVD市場にこれから参入しようとしている企業
医療機器・IVDを海外に輸出しようとしている企業
FDA査察準備、MDAPを受審しようと準備している企業
既にFDA査察を終え、指摘対応に苦慮している企業
受講料: ¥35,000( テキスト含む )
割 引: ¥30,000( テキスト含む )
1社2名様以上受講される場合
1日コース:午後開催される『 QMS購買管理コース 』を引き続き受講される方
受講料: ¥52.000-( 1日、テキスト代、昼食代含む )
割 引: ¥48.000-
1社2名様以上ご参加の場合
定 員: 24名
出席者全員に教育記録として利用できる修了書をお渡しします
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡いたします。
2名様以上ご参加の場合、お名前とローマ字読み、メールアドレスを連絡欄にご記入下さい。
持参品: 弊社QSR翻訳本第三版をお勧めします。お持ちでない方はこちらからお求め頂けます。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.FDA規制、査察の最新情報と傾向分析
規制当局の警告書分析と傾向
2.医療機器規制・規格の理解
適用規制・規格
品質計画( 付帯サービスプロセス )
プロセスアプローチKPI
タートルチャート
3.付帯サービスの目的の理解
付帯サービスの目的
QSRとISO、省令から理解する
言葉の定義
4.サービス設計
ライフサイクルサービス活動の設計
820.30 の適用
コンカレント設計とは
設計アウトプット(サービス向け)
5.ライフサイクルマネジメント
PM1:製品開発の上流から参画しサービス設計実施
ライフサイクルマネジメントサービス活動
設計へのインプット
サービス要求仕様書
サービス活動のリモート化
M2 :サービス診断ツールの検証
サービス効率化に欠かせない診断ツール
診断ツールの検証
M3 :サービスマニュアル検証とバリデーション
マニュアルの検証とバリデーション
サービストレーニング
インスタレーション規制の理解
M4 :パーツハンドリング
サービスパーツの保管
S−BOM
劣化製品の取扱
6.有害事象報告(MDR)
サービス報告と苦情
MDRとは
MDR提出判断とフロー
7.統計的傾向監視
統計的分析
CAPA
上流へのフィードバック
QMS 購買管理コース
〜 SCM でQCDに貢献する購買プロセスの本質 〜
21CFR Part 820・ISO13485・ISO9001
このコースの売り上げ金の一部は人道支援活動に寄付されます
募集を締め切らせて頂きました
☆ 開催概要 ☆
購買プロセスは言われた通りにモノを買っていればよいというものではなく、サプライチェーン全体を俯瞰し、製品企画時の上流から積極的にコンカレントで参画、最も効率よいQCDを提案することこそ会社に貢献するプロセスとして価値があります。その為には、戦略的な組織体制とフレームを構築する必要があり、開発購買や情報購買と言った戦略的なフレームは早期の製品ローンチを実現します。また、アッセンブルラーが多くなった昨今、行政当局も購買プロセスに多くの査察時間を費やすようになりました。自社と同じシステムで管理する必要性が高まり、フレームと管理能力が要求されます。FDA QSRのみならず、ISOの基本9001の要求も加味しながら理解を深めます。
オンサイトでも可能です。お問い合わせはこちらから。
日 時:2019年7月8日(月) 13:00 〜 16:30 半日コースです
会 場:全国町村会館(東京・永田町) 永田町駅から徒歩1分( 3番出口 )
講 師:クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
ODM、OEMとして設計・製造を受託している企業
受講料: ¥42.000-( 午後半日、テキスト代含む )
割 引: ¥38.000-
1社2名様以上ご参加の場合
1日コース:午前中開催される『 付帯サービスコース 』を引き続き受講される方
受講料: ¥52.000-( 1日、テキスト代、昼食代含む )
割 引: ¥48.000-
1社2名様以上ご参加の場合
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
持参品: 弊社QSR翻訳本第三版をお勧めします。お持ちでない方はこちらからお求め頂けます。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
A.FDA規制、査察の最新情報と傾向分析
1.適用規制・規格
FDA QSR,ISO13485,9001の要求とは
プロセスアプローチでインとアウトを理解する
2.購買管理の目的
規格が示す購買管理の目的
製品、サービス購買とは
4つの購買範囲とは
3.用語の定義
製品、サービスとは
プロバイダー、サプライヤー、コントラクターとは
4.購買管理
GHTF( IMDRF )ガイダンスのフロー
5.購買方針
購買方針がなければ購買プロセスは成り立たない
6.戦略・開発購買
サプライチェーン全体を俯瞰した戦略
開発スピードを上げ原価削減、購買活動のスピードを上げある
ベストプラクティスの収集と技術の取り込み
7.購買計画
製品企画段階から参画
供給する製品、サービスの特定
技術及びプロセス情報の特定
供給者候補の特定
リスク、管理方法の特定
8.供給者候補選定
供給者ビジネス、運用能力評価
供給者候補選定
9.供給者選定
評価選定基準の策定
供給者候補の能力評価
供給者の承認
10.管理方法の取り決め
購買情報とは
管理方法
11.変更時対応
変更管理規定
4Mと変化点管理
12.納期、測定、監視
製品、サービスの検証
不適合の手順
供給者の監視
製品の受領、出荷場、冷蔵室、NCエリア
カーゴ内温度バリデーション
13.Feedback, Communication
CR
SCAR
供給者評価シート
14.Supplier Audit
リスクベース監査
5つの監査、評価エリア
情報購買、戦略上重要な監査チーム
( 内容が変更になる場合もございますが大きな変更はございません。)

2018_10_PC
FDA 設計管理2日間コース Rev.22
〜 リスクをヘッジしコンプライアンスを堅持するフレームワーク 〜
21CFR Part 820.30 / 820.75
このコースの売上金の一部は人道活動支援に寄付されます
12月開催、QSR内部監査員コースご出席要件コースです
( オンサイト及び過去弊社コースを受講された方は要件を満たします )
定員オーバーのため募集を締め切らせて頂きました
次回(10月)のご参加をお待ちしています
☆ 開催概要 ☆
ほとんどの日本企業に於ける医療機器開発・設計管理の仕組みは世界標準にほど遠く、これはグローバルに進出しようとする企業のみならず、患者の命を預かる全ての医療機器メーカーにとって大きなリスクを伴います。米国FDAは自らの査察で、医療機器品質不良の根本原因は上流側の設計管理にあると査察の結果から統計的に見抜いており、それはクオリス・イノーバの10年以上にわたるGAP監査でも明らかです。そこで本コースでは、グローバルな開発、設計管理を米国FDA「設計管理ガイダンス」及び欧米で取り入れられている開発フレームワークを元に体系的に解説、また、品質不良を発生させない仕組み(ロバスト設計)をタグチメソッドと設計CAPAを用いて解説します。 2日目はプロセスバリデーションの本質、工程内試験に頼らない均一で高品質な製品を一貫して製造する為、パラメータによる製造方法を実現する工程設計について解説します。 ”規制” だから、ではなく、設計品質を高めることにより患者さんの命を守り、企業にとっても大きな潜在的利益を得られるという大原則は、エンジニアの方々から多くの共感と支持を頂いてきたクオリス・イノーバの最も人気のある価値あるコースです。 このコースはオンサイトでも実施しております。お問い合わせはこちらから。
日 時: 2019年6月17日(月曜)、18日(火曜) 10:00〜16:30
会 場: 全国町村会館(東京・永田町) 永田町駅から徒歩1分( 3番出口 )
講 師: クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象: 医療機器・IVD市場にこれから参入しようとしている企業
医療機器・IVDを海外に輸出しようとしている企業
FDA査察準備、MDAPを受審しようと準備している企業
既にFDA査察を終え、指摘対応に苦慮している企業
受講料: ¥99,000( テキスト・昼食含む )
割 引: ¥85,000( テキスト・朝食含む )
1社2名様以上受講される場合
定 員: 24名
出席者全員に教育記録として利用できる修了書をお渡しします
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡いたします。
2名様以上ご参加の場合、お名前とローマ字読み、メールアドレスを連絡欄にご記入下さい。
持参品: 弊社QSR翻訳本第三版をご持参下さい。お持ちでない方はこちらからお求め頂けます。
ご注意: 本コースは、品質システムの一部を解説するため、システム全体の知識がないと理解できません。本コース受講前に5月開催QSR2日間コースの受講をお勧めします。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1日目(セミナー内容)
1.設計品質の本質的価値
TQMに欠かせないマネジメントレビューの重要な役割
設計環境、開発リソースの見極め
要員教育、開発リソースの見極め
力量表と教育、訓練計画
2.820.30 (a) 総則
繰り返される設計品質に起因する製品不良
ISOとQSRの本質的な相違
理解すべき規制・規格・ガイダンス
FDAが望んでいるTQMの本質、品質工学に基づくロバスト設計
医療機器の設計手法( Water fall model )
一貫性を問うFDA査察
3.820.30 (b) 設計及び開発計画
医療機器開発ライフサイクルマネジメント(LCM)
ライフサイクルリスクアセスメント
タグチメソッドの開発方式によるフロントローディング手法
PM、PLの役割
カスタマーニーズを要求仕様に変換するQFD
設計品質確立の重要な要素、コンカレントエンジニアリング(CE)
設計計画書とは
4.820.30 (c) 設計インプット
ロバスト設計(タグチメソッド)による未然防止設計
要求仕様書から設計仕様書への変換
曖昧なニーズのスペックへの変換
重要な設計要素トレーサビリティーマトリックス(TMX)
ラベリング、包装設計のインプット
5.820.30 (d) 設計アウトプット
入力のバラツキを押さえたアウトプットの確認
設計アウトプットを定義しアウトプット文書を明確にする
E-BOM の役割
製造仕様書とは
図面発行
DMRとトレーサビリティー
6.820.30 (e) 設計レビュー
問題の早期発見、解決が目的でセレモニーで終わらせない
設計審査で設計のロバストネスを確認
設計審査とテクニカルレビュー
審査者の力量
7.820.30 (f) 設計検証
設計検証と設計バリデーションの相違
インプットからアウトプットまでのトレーサビリティー
エビデンスに基づく設計
設計検証計画に必要な要素
設計検証レポートはデータの信頼性を確保
8.820.30 (g) 設計バリデーション
設計バリデーションの手法
社内、臨床評価の相違と記録
ソフトウェアのバリデーションとVモデル
3種類のバリデーションの相違
9.820.30 (h) 設計移管 → 第2日目(工程設計)
10.820.30 (i ) 設計変更
DHF / DMR / DHR の定義
識別、ファイルの基本
文書体系と設計標準
2日目(セミナー内容)
設計移管と工程設計
1.なぜ工程バリデーションを実施するのか
プロセスバリデーションの目的とその本質
プロセスバリデーションの効果
工程管理、監視の目的と本質
医療機器規制・規格の理解
2.バリデーションすべき工程とは
バリデーションすべき工程の識別
検証工程とバリデーション工程の相違
設備バリデーションと工程バリデーション
バリデーション工程例
3.バリデーションの種類
バリデーションの種類
設備バリデーションと劣化パラメータ
コンピューターシステムバリデーション(CSV)
4.工程設計とバリデーションステップ
VMP:バリデーションマスタープラン
DQ: 設計時適格性評価
IQ: 据付時適格性評価
OQ: 稼働時適格性評価
PQ: 製造時適格性評価
PPQ: プロセス街道性評価
バリデーション文書化
プロセスバリデーションの明確化
自動化工程の明確化
CSVカテゴリーの明確化
ソフトウェアのバリデーションの基本
リスクベースCSV
5.820.30 (h) 設計移管
820.75 プロセスバリデーション
820.72 検査、測定および試験装置
設計移管とプロセスバリデーションの関係
EV、PV、検査測定装置の妥当性とは
ミニスケールとフルスケール
工程設計とPTMX
6.バリデーションの実践
バリデーションマスタープラン(VMP)
バリデーション工程の識別
バリデーション計画
設備リストとバリデーション工程リスト
チームビルディング
DQP/DQR
IQP/IQR
OQP/OQR
PQP/PQR
PPQP/PPQR
7.バリデーションアウトプット
工程能力
再バリデーション設定
設備メンテナンス手順
( 内容が変更される場合がございます )

2018_11 設計管理2日間コース
FDA QSR2日間コース Rev.22
〜 医療機器・IVDに求められる規制要求事項の本質を学ぶ 〜
21CFR Part 820
このコースの売り上げ金の一部は人道支援活動に寄付されます
ISO13485 との比較をより詳細に行いシステム構築に最適です
12月開催 FDA 内部監査員コースの出席される場合の要件コースです
設計管理コース、購買管理コースを受講される方も本コースを受講されることをお勧めします
定員オーバーの為募集を締め切りました
次回(9月)のご参加をお待ちしております!
☆ 開催概要 ☆
* FDA 品質システム規制 QSR 対訳本( 第3版 )付き
グローバルに進出しようとする企業にとって 米国は最も大きなマーケットであり、米国市場での成功がグローバルを制するといっても過言ではありません。しかし、米国に上市するには医療機器規制QSRに基づくFDAの査察は避けられません。( MDSAPを受審される企業も必須 )また、医療機器を国内で販売する際にもFDA QSRのシステムフレームワーク、エビデンスベースの考え方を学び取り入れる事は、製品品質確保につながり、リスクの低減、新しいISO13485にも準拠し、特に受託設計、製造会社 ( ODM、OEM ) では会社のプレゼンスを高め、サプライヤ−としての評価を高めます。 クオリス・イノーバのQSRコースは、多くのFDA査察準備、対応の経験や品質管理の経験から、FDA規制のみならず医療機器開発・製造の本質を鋭く説き、多くの企業の皆様の共感を集め支持されて参りました。米国に進出する企業のみならず、医療機器・IVD を開発しようとしている企業の皆様のマインドが大きく変わる価値あるクオリス・イノーバのメインコースです。
オンサイトでも可能です。お問い合わせはこちらから。
日 時:2019年5月13日(月)、14日(火) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
ODM、OEMとして設計・製造を受託している企業
受講料: ¥99,000( 2日分、テキスト・対訳本(3版)・昼食代含む )
割 引: ¥85,000
1社2名様以上ご参加の場合
特 割: 企業で行うプロジェクトの後、継続研修などでご利用の場合、
特別割引を適用させて頂きますのでメールでご相談下さい。
この場合、正規料金をお支払いの方が優先となりご希望に添えない場合が
ございますので予めご了承ください。
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
第1日目 セミナー内容
1.コンプライアンスとインテグリティー
患者の命と会社の危機管理
データインテグリティー
トップのリーダーシップ
2.行政によるミッションの相違
3.米国法規制の理解
4.医療機器医規制の理解
5.なぜISOではFDA査察にパスしないのか
6.QSITアプローチとは
7.規制を正しく理解する要素:定義
820.20 経営者の責任
経営者がリードするTQMが基本
マネジメントレビューの本質、プロセスアプローチ
プロセスを常に改善する
820.22 品質監査
効果的な監査手法と監査報告のあり方
内部監査はプロセスの異常を見極める
内部監査員はなぜ異常を見極められないのか
820.25 要員
教育の重要性:教育の手法と記録、力量表、リソースとは
教育と訓練の相違、アウェアネストレーニングの実施と訓練の重要性
820.30 設計管理( 概要のみ:6月開催 設計監理2日コースで詳細解説 )
ライフサイクル製品開発手法、ウオーターフォールモデル、
フロントローディング手法、ロバスト設計
設計検証と設計バリデーションの相違
今までISO になかった設計移管、工程設計とは
設計CAPAとは
820.40 文書管理
グロバールに通用する文書管理
品質を担保するSOPの作成方法
820.50 購買管理
開発購買とその役割、購買プロセス
3つの購買(製品、サービス、一般)と購買製品検証
新規サプライヤ評価と継続評価
購買情報とは
820.60 識別
混同を防止する為の識別方法とは
820.65 トレーサビリティー
体内埋込型機器だけではなく、変化点を知り品質を高める
820.80 アクセプタンスアクティビティー
3つのアクセプタンスを理解する
Receiving, In-process, and Finished device acceptance
820.86 アクセプタンス状態
状態表示する意味とは
820.90 不適合品
不適合品の処理プロセス
FDAはなぜNCを見たがるのか
グローバル用語を使用し、CAPAとのリンクを示す
820.120 機器のラベリング
リコールが一番多く査察で注目されるラベリングの出納管理
ラベリングの設計とは
ラベルとラベリングの相違
820.130 機器の梱包
梱包箱を見ればどんな設計を行っていたかが判る
設計依託先はISOで十分か?
820.140 取扱い
混同・劣化・損傷・汚染を発生させないためのリスク管理
820.150 保管
リスクベース保管管理
820.160 流通
ユーザーに届けるまでの製品設計
リスクベース流通管理
820.170 据付け
インスタレーション手順書のV&V
820.180 記録
全ての活動が根拠に基づくエビデンスで保証されるための重要な活動
Objective Evidence を示すための記録管理とは
820.181 機器原簿(DMR)
DMRを定義し識別する
DHF,DMR,DHRの相違
820.184 機器履歴簿(DHR)
製造記録の記載方法
コンプライアンス、製品品質を立証するため重要なDHR
820.186 品質システム記録
DHF,DMR,DHR以外の品質記録とは
820.200 付帯サービス
サービス記録の分析とMR報告,MDRの判定
820.250 統計的手法
TQMを進めていく上で最も重要な要素
品質改善に欠かせない統計的手法
第2日目 セミナー内容
820.70 生産及び工程管理
工程設計と工程V&V
( 概要のみ:6月開催 設計監理2日コースで詳細解説 )
工程のリスクマネジメント(PFMEA)
工程を監視、管理する工程パラメータとは
要員、環境、汚染管理
設備の妥当性とは
保守と検査の相違
副資材の定義と管理
820.72 検査・測定及び試験装置
意図した用途への適合性、測定器の精度管理とは
測定器の精度管理と救済処置
820.75 工程の妥当性確認
QSR,医療機器規制の本質、工程バリデーションとは
( 概要のみ:6月開催 設計監理2日コースで詳細解説 )
TQMが目指すべき試験レスとは
バリデーションの手法( VMP,DQ、IQ、OQ、PQ、PPQ )
設備バリデーションとは
CSV( コンピュータシステムバリデーション )とは
820.198 苦情ファイル
査察のキーポイント、苦情管理の目的を理解する
苦情処理とMDR( Part 803 )、CAR( Part 806 ) の関わり
( 概要のみ:MDRコースで詳細解説 )
820.100 是正処置及び予防処置( CAPA )
FDA警告書で最も多い指摘CAPAの原則を理解する
品質システムの本質、プロセスアプローチの重要な要素
修正、是正、予防の相違を正確に理解しISOマインドを変える
CAPA7ステップを理解する
CAPAの効果(再発防止)を出す仕組み
MRで監視すべきCAPAプロセス
( 概要のみ:CAPAコースで詳細解説 )
( 内容が変更になる場合もございますが大きな変更はございません。)
2018_10 QSR Seminar 様子

CAPAコース Rev.18
〜 指摘の最も多いCAPAの理解と問題の予防と再発防止の仕組み 〜
このコースの売上金の一部は人道支援に寄付されます
定員をオーバーした為受付を締め切りました。
次回のご参加をお待ちしております。
* コース概要 *
CAPAプロセスを確認するだけで、その会社の品質システムや品質に対する考え方がよくわかります。CAPAプロセスに問題があれば、品質マネジメントシステムの適切性、妥当性はなく、特にマネジメントレビューがまともに運用されているとは到底思われません。品質マネジメントシステムの原則はプロセスアプローチ、PDCAをまわす為、マネジメントレビューのインプットでプロセスを監視し、改善ツールであるCAPAでプロセスを改善します。当たり前のことがシステムとして運用されていないことを理解している規制当局は、CAPAプロセスにフォーカスして確認しようとします。QMSが機能していないことは昨今の品質に関わる事件から明らかで、医療機器・IVDの場合、命に関わる大問題です。本コースでは今までの内容を大きく変更し、プロセスアプローチの手法に重点を置き、事例を元にCAPAの本質を理解して頂きます。 このコースは受講者と同じ目線の御社事例を用いることでより理解が高まるオンサイトセミナーが可能です。お問合せはこちらから。
日 時:2019年3月4日(月曜)10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 ) ( 永田町駅から徒歩1分 )
講 師:クオリス・イノーバ株式会社代表取締役社長 木村 浩実
対 象:医療機器、IVD、医薬品等に限定せず、CAPAプロセスを学びたい企業
受講料: ¥50,000( テキスト代・昼食含む )
割 引: ¥40,000( テキスト代・昼食含む )
1社2名様以上ご参加の場合
特 割:弊社プロジェクトを実施中の場合や、継続研修をご希望の場合、ご負担軽減のためさらに
割引をさせて頂きますのでお問い合わせください。 この場合、正規料金をお支払いのお
客様が優先され、ご出席できない場合がございますのでご了解ください。
再受講:空席がある場合、特別な料金で再受講可能ですのでお問い合わせください。
併せて受講された日もご記入ください。
定 員:20名様 2月26日締め切り
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込:セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールで連絡いたします。
複数名ご参加の場合、同様の情報をご記入下さい。 割引料金でご案内いたします。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.なぜFDAはCAPAに注目するのか
2.なぜ品質問題は再発するのか
3.規制・規格を正しく理解する
4.プロセスアプローチの基本
プロセス関連図
タートルチャート
5.TQMの仕組みが不可欠
マネジメントレビュー
PDCAをまわす
プロセス改善ツール
6.用語の正しい理解
修正、是正、予防の相違
逸脱、不適合、逸脱許可と特別採用の相違
修正、手直し、修理の相違
7.CAPAプロセスの IN・OUT
CAPAタートルチャート
不適合処理プロセス
苦情処理プロセス
CAPAプロセス
MDRプロセス
リコールプロセス
8.直接原因と根本原因を理解する
直接原因と根本原因の相違
根本原因の改善がプロセスアプローチの本質
9.予防プロセス
品質管理の原則、予防活動の理解
10.エスカレーション
CAPAエスカレーション
CAPAを上手に運用するための仕組み
11.CAPA7ステップ
Step 1 問題を検出し明確にする
Step 2 問題の原因を詳しく調査する
Step 3 根本原因の特定
Step 4 処置の特定と検証または妥当性確認
Step 5 予防または是正する為の処置
Step 6 処置の結果を文書化
Step 7 有効性の確認
12.CARの実施
13.設計CAPA
14.CAPAマネジメント
マネジメントレビューの運用方法
15.品質問題の事例に学ぶ
Warning Letter Case Study
* 内容(項目)が変更される場合があります

2018_3 CAPA コース
FDA MDR&CAR コース Rev.4
〜 指摘の多い有害事象報告・リコール規制の理解とプロセス構築 〜
21CFR : Part 803, 806
このコースの売り上げ金の一部は人道支援活動に寄付されます
☆ 開催概要 ☆
2016年FDA査察レポートの解析からワースト4位(全体の約10%)に急浮上しているMDR(有害事象報告)、日本でも Warning Letter (警告書)を受けているほとんどの企業がMDRの指摘を受けています。昨今のデータ偽装事件を受け、FDAはこれらにフォーカスし厳しく査察を行っています。( 2018FDAレポート)。これはMDR報告の判断に迷ったら報告するという米国の文化とは異なり、日本のメーカーは情報を積極的に発信しない、隠すという古い会社の文化が根強くあることが要因の一つですが、そもそも規制を理解せず正しい判断を行っていないことが大きな原因です。医療機器を作っているメーカーは、患者さんの命を守るという社会的な責任があります。会社を守っているようでも、結果的に会社が倒産に追い込まれることもある非常にリスクの高いプロセスであることを理解すべきです。それには規制の正しい理解、規制を守る仕組みと組織が必要です。このセミナーでは、CAPAサブシステム(苦情、不適合、CAPA)の各プロセスとリンクし、日本のメーカーの Warning Letter (警告書)を引き合いに正しいMDRプロセスと関連するCAR(リコール)プロセスを構築することを理解します。このコースはオンサイトでも実施しています。お問い合わせはこちらから。
日 時:2019年1月28日(月曜)13:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:既に医療機器・IVDを米国に輸出している企業
これから医療機器・IVDを米国輸出する準備を行っている企業
既にFDA査察を終え対応に困っている企業
受講料: ¥42,000-
割 引: ¥38,000-( 1社2名様以上受講される場合 )
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.規制を理解する
事例と統計からみる規制違反
理解すべき規制・規格・ガイダンス
eMDR規制
規制が求めるあるべき組織と力量
2.有害事象報告( MDR )
規制の目的、重要性
MDRとして報告すべき事象とは
5日報告、30日報告とは
業態(使用施設、輸入業者、製造業者)毎の報告内容
3.輸入業者との関係
輸入業者のMDRプロセス把握
プロセスの共有と規制が求める契約
eMDR登録
4.MDRプロセス
P820に基づく苦情処理プロセス(グローバル)
P820に基づく不適合処理プロセス
規制に基づくMDRプロセスとは
5.警告書から理解すべき注意点とプロセスへの展開
日本の Warning Letter ( 警告書 )の事例から理解する
警告書から理解すべき注意点とプロセスへの展開
プロセス構築後のレトロスペクティブな是正とは
6.CAR( リコール )プロセス
改修及び回収(CAR)
用語の定義を理解する
報告義務のないCARとは
CARプロセス
ディシジョンツリー
5.警告書から理解すべき注意点とプロセスへの展開
日本の Warning Letter ( 警告書 )の事例から理解する
警告書から理解すべき注意点とプロセスへの展開
*コース内容が一部変更される場合がございますが大きな変更はございません。

2018 MDR/CAR
FDA QSR 内部監査員コース Rev.8
〜 QSR 対応内部監査員養成のための社内資格認定2日間コース 〜
* よりプロセスアプローチを理解するコースに改定 *
このコースの売り上げ金の一部は人道支援活動に寄付されます
☆ 開催概要 ☆
不適合を出さないようプロセスに潜在する問題を未然に抽出し、マネジメントレビューでトップの指示のもとプロセス改善を常に行う。これがTQMの有るべき姿であり規制当局が求めている姿です。一方、現実はプロセスを監視し傾向管理を行っておらず、よく訓練されていないにわか監査員が規制・規格で要求されているからといって片手間に監査を行っているだけ。これは医療機器の場合致命的で、リコールで何十億の損失を出すだけではなく、人の命に関わる重大な問題に発展し会社の存続まで危ぶまれます。このコースでは、ISO内部監査員の資格を持つ方々がQSRの内部監査が行えるよう社内資格を付与するための社内資格認定コースとして位置づけられます。一般に行われているISOの内部監査手法とは異なる、付加価値を生む予防対策としての監査手法とノウハウを学びます。( このコースを受講した後、オンサイトでクオリス・イノーバによる内部監査のOJTを行うとより効果的です )
日 時:2018年12月3日(月曜)、4日(火曜) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:すでに弊社主催下記コースをオープン、オンサイト問わず受講された方限定です。
* FDA GMP QSR 2日間コース
* FDA 設計管理2日間コース
このコースはQSRの基礎知識が無いと受講できず、弊社のコースを受講されていない場合
異なる解釈により内容をよく理解出来ません。 何卒ご理解をお願い申し上げます。
受講料: ¥99,000( テキスト、昼食代含む、内税 )
このコースには割引がございません。
定 員: 18名限定 満席になり次第締切ます
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
過去に受講された弊社QSR、設計管理各コースの受講日を記載して下さい。
オンサイトで受講された場合でも構いません。
折り返し、会場、お振込先情報をメールでご連絡致します。
セミナーに参加するための宿題を追って発送し致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.内部監査の目的を理解する
規制・規格が求めていること
患者の命を優先する監査とは
企業の付加価値を高める為には
2.内部監査員に求められる力量
内部監査員とリーダーに求められる力量
内部監査員訓練
マネジメントチームのカウンセラー
3.プロセスアプローチ
CAPAの理解とCAPAアプローチ
プロセスのR&R( 役割と責任 )を明確にする
プロセスを常に測定し改善する
内部監査はプロセスに注目
4.監査判定基準
チェックリストと数値化
プロセス毎に数値化
FDA査察と内部監査判断基準の相違を知る
5.監査を計画する
マネジメントレビューと品質会議からの指示
リスクベース&プロセスアプローチによる監査計画
プロセス管理パラメートと傾向分析結果
是正監査と予防監査の相違
監査は短期集中型で実施
監査員レビュー会のノウハウ
監査報告書記載のポイント
6.内部監査レポート
トップマネジメントに対する報告
プロセスの不足、改善点を報告
規制に対するコンプライアンス状況の報告
トップマネジメントのアクション
7.監査の目のつけどころ( QSIT )
QSITに基づく監査手法と勘所
プロセスアプローチの手法を全員が理解(宿題あり)
1.経営者による管理
2.設計管理
3.CAPA
MDR報告
CAR
MDトラッキング
4.P&PC
5.滅菌工程
+6.各プロセス
*内容は変更される場合がございます*
2017_12_Internal auditor
FDA 設計管理2日間コース Rev.21
〜 リスクをヘッジしコンプライアンスを堅持するフレームワーク 〜
21CFR Part 820.30
定員オーバーのため募集を締め切りました
次回のご参加をお待ちしております
このコースの売上金の一部は人道活動支援に寄付されます
12月開催、QSR内部監査員コースご出席要件コースで、
10月開催QSR2日間コースも出席要件となりますので併せてご出席下さい。
( オンサイト及び過去弊社コースを受講された方は要件を満たします )
☆ 開催概要 ☆
ほとんどの日本企業に於ける医療機器開発・設計管理の仕組みは世界標準にほど遠く、これはグローバルに進出しようとする企業のみならず、患者の命を預かる全ての医療機器メーカーにとって大きなリスクを伴います。米国FDAは自らの査察で、医療機器品質不良の根本原因は上流側の設計管理にあると査察の結果から統計的に見抜いており、それはクオリス・イノーバの10年以上にわたるGAP監査でも明らかです。そこで本コースでは、グローバルな開発、設計管理を米国FDA「設計管理ガイダンス」及び欧米で取り入れられている開発フレームワークを元に体系的に解説、また、品質不良を発生させない仕組み(ロバスト設計)をタグチメソッドと設計CAPAを用いて解説します。 2日目はプロセスバリデーションの本質、工程内試験に頼らない均一で高品質な製品を一貫して製造する為、パラメータによる製造方法を実現する工程設計について解説します。 ”規制” だから、ではなく、設計品質を高めることにより患者さんの命を守り、企業にとっても大きな潜在的利益を得られるという大原則は、エンジニアの方々から多くの共感と支持を頂いてきたクオリス・イノーバの最も人気のある価値あるコースです。 このコースはオンサイトでも実施しております。お問い合わせはこちらから。
日 時: 2018年11月19日(月曜)、20日(火曜) 10:00〜16:30
会 場: 全国町村会館(東京・永田町) 永田町駅から徒歩1分( 3番出口 )
講 師: クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象: 医療機器・IVD市場にこれから参入しようとしている企業
医療機器・IVDを海外に輸出しようとしている企業
FDA査察準備、MDAPを受審しようと準備している企業
既にFDA査察を終え、指摘対応に苦慮している企業
受講料: ¥99,000( テキスト・昼食含む )
割 引: ¥85,000( テキスト・朝食含む )
1社2名様以上受講される場合
定 員: 24名
出席者全員に教育記録として利用できる修了書をお渡しします
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡いたします。
2名様以上ご参加の場合、お名前とローマ字読み、メールアドレスを連絡欄にご記入下さい。
持参品: 弊社QSR翻訳本第三版をご持参下さい。お持ちでない方はこちらからお求め頂けます。
ご注意: 本コースは、品質システムの一部を解説するため、システム全体の知識がないと理解できません。本コース受講前に10月開催QSR2日間コースの受講をお勧めします。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1日目(セミナー内容)
1.設計品質の本質的価値
TQMに欠かせないマネジメントレビューの重要な役割
設計環境、開発リソースの見極め
要員教育、開発リソースの見極め
力量表と教育、訓練計画
2.820.30 (a) 総則
繰り返される設計品質に起因する製品不良
ISOとQSRの本質的な相違
理解すべき規制・規格・ガイダンス
FDAが望んでいるTQMの本質、品質工学に基づくロバスト設計
医療機器の設計手法( Water fall model )
一貫性を問うFDA査察
3.820.30 (b) 設計及び開発計画
医療機器開発ライフサイクルマネジメント(LCM)
ライフサイクルリスクアセスメント
タグチメソッドの開発方式によるフロントローディング手法
PM、PLの役割
カスタマーニーズを要求仕様に変換するQFD
設計品質確立の重要な要素、コンカレントエンジニアリング(CE)
設計計画書とは
4.820.30 (c) 設計インプット
ロバスト設計(タグチメソッド)による未然防止設計
要求仕様書から設計仕様書への変換
曖昧なニーズのスペックへの変換
重要な設計要素トレーサビリティーマトリックス(TMX)
ラベリング、包装設計のインプット
5.820.30 (d) 設計アウトプット
入力のバラツキを押さえたアウトプットの確認
設計アウトプットを定義しアウトプット文書を明確にする
E-BOM の役割
製造仕様書とは
図面発行
DMRとトレーサビリティー
6.820.30 (e) 設計レビュー
問題の早期発見、解決が目的でセレモニーで終わらせない
設計審査で設計のロバストネスを確認
設計審査とテクニカルレビュー
審査者の力量
7.820.30 (f) 設計検証
設計検証と設計バリデーションの相違
インプットからアウトプットまでのトレーサビリティー
エビデンスに基づく設計
設計検証計画に必要な要素
設計検証レポートはデータの信頼性を確保
8.820.30 (g) 設計バリデーション
設計バリデーションの手法
社内、臨床評価の相違と記録
ソフトウェアのバリデーションとVモデル
3種類のバリデーションの相違
9.820.30 (h) 設計移管 → 第2日目(工程設計)
10.820.30 (i ) 設計変更
DHF / DMR / DHR の定義
識別、ファイルの基本
文書体系と設計標準
2日目(セミナー内容)
設計移管と工程設計
1.なぜ工程バリデーションを実施するのか
プロセスバリデーションの目的とその本質
プロセスバリデーションの効果
工程管理、監視の目的と本質
医療機器規制・規格の理解
2.バリデーションすべき工程とは
バリデーションすべき工程の識別
検証工程とバリデーション工程の相違
設備バリデーションと工程バリデーション
バリデーション工程例
3.バリデーションの種類
バリデーションの種類
設備バリデーションと劣化パラメータ
コンピューターシステムバリデーション(CSV)
4.工程設計とバリデーションステップ
VMP:バリデーションマスタープラン
DQ: 設計時適格性評価
IQ: 据付時適格性評価
OQ: 稼働時適格性評価
PQ: 製造時適格性評価
PPQ: プロセス街道性評価
バリデーション文書化
プロセスバリデーションの明確化
自動化工程の明確化
CSVカテゴリーの明確化
ソフトウェアのバリデーションの基本
リスクベースCSV
5.820.30 (h) 設計移管
820.75 プロセスバリデーション
820.72 検査、測定および試験装置
設計移管とプロセスバリデーションの関係
EV、PV、検査測定装置の妥当性とは
ミニスケールとフルスケール
工程設計とPTMX
6.バリデーションの実践
バリデーションマスタープラン(VMP)
バリデーション工程の識別
バリデーション計画
設備リストとバリデーション工程リスト
チームビルディング
DQP/DQR
IQP/IQR
OQP/OQR
PQP/PQR
PPQP/PPQR
7.バリデーションアウトプット
工程能力
再バリデーション設定
設備メンテナンス手順
( 内容が変更される場合がございます )

2018_6 設計管理2日間コース
QMS 購買管理コース
〜 医療機器・IVDに求められる規制要求事項の本質を学ぶ 〜
21CFR Part 820・ISO13485・ISO9001
定員オーバーのため募集を締め切りました
次回のご参加をお待ちしております
このコースの売り上げ金の一部は人道支援活動に寄付されます
このコースの内容を理解するために
QSR2日間コースでシステムの基礎を学ばれることをお勧めします
☆ 開催概要 ☆
購買プロセスは言われた通りにモノを買っていればよいというものではありません。サプライチェーン全体を俯瞰し、製品企画時に積極的にコンカレントで参画、最も効率よいQCDを提案することこそ会社に貢献するプロセスとして価値があります。その為には、戦略的な組織体制とフレームを構築する必要があり、開発購買や情報購買と言った戦略的なフレームは早期の製品ローンチを実現します。さらに、アッセンブルラーが多くなった昨今、行政当局も購買プロセスに多くの査察時間を費やすようになりました。自社と同じシステムで管理する必要性が高まり、フレームと管理能力が要求されます。FDA QSRのみならず、ISOの基本9001の要求も加味しながら理解を深めます。
オンサイトでも可能です。お問い合わせはこちらから。
日 時:2018年10月29日(月) 13:00 〜 16:30 半日コースです
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
ODM、OEMとして設計・製造を受託している企業
受講料: ¥42.000-( 午後半日、テキスト代含む )
割 引: ¥38.000-
1社2名様以上ご参加の場合
特 割: 企業で行うプロジェクトの後、継続研修などでご利用の場合、
特別割引を適用させて頂きますのでご相談下さい。
この場合、正規料金をお支払いの方が優先となりご希望に添えない場合が
ございますので予めご了承ください。
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
A.FDA規制、査察の最新情報と傾向分析
1.適用規制・規格
FDA QSR,ISO13485,9001の要求とは
プロセスアプローチでインとアウトを理解する
2.購買管理の目的
規格が示す購買管理の目的
製品、サービス購買とは
4つの購買範囲とは
3.用語の定義
製品、サービスとは
プロバイダー、サプライヤー、コントラクターとは
4.購買管理
GHTF( IMDRF )ガイダンスのフロー
5.購買方針
購買方針がなければ購買プロセスは成り立たない
6.戦略・開発購買
サプライチェーン全体を俯瞰した戦略
開発スピードを上げ原価削減、購買活動のスピードを上げある
ベストプラクティスの収集と技術の取り込み
7.購買計画
製品企画段階から参画
供給する製品、サービスの特定
技術及びプロセス情報の特定
供給者候補の特定
リスク、管理方法の特定
8.供給者候補選定
供給者ビジネス、運用能力評価
供給者候補選定
9.供給者選定
評価選定基準の策定
供給者候補の能力評価
供給者の承認
10.管理方法の取り決め
購買情報とは
管理方法
11.変更時対応
変更管理規定
4Mと変化点管理
12.納期、測定、監視
製品、サービスの検証
不適合の手順
供給者の監視
製品の受領、出荷場、冷蔵室、NCエリア
カーゴ内温度バリデーション
13.Feedback, Communication
CR
SCAR
供給者評価シート
14.Supplier Audit
リスクベース監査
5つの監査、評価エリア
情報購買、戦略上重要な監査チーム
( 内容が変更になる場合もございますが大きな変更はございません。)
FDA QSR2日間コース Rev.21
〜 医療機器・IVDに求められる規制要求事項の本質を学ぶ 〜
21CFR Part 820
定員オーバーとなりましたので募集を締め切りました
次回のご参加をお待ちしております
このコースの売り上げ金の一部は人道支援活動に寄付されます
12月開催 FDA 内部監査員コースの出席される場合の要件コースです
設計管理コース、購買管理コースを受講される方も本コースを受講されることをお勧めします
☆ 開催概要 ☆
* FDA 品質システム規制 QSR 対訳本( 第3版 )付き
グローバルに進出しようとする企業にとって 米国は最も大きなマーケットであり、米国市場での成功がグローバルを制するといっても過言ではありません。しかし、米国に上市するには医療機器規制QSRに基づくFDAの査察は避けられません。( MDSAPを受審される企業も必須 )また、医療機器を国内で販売する際にもFDA QSRのシステムフレームワーク、エビデンスベースの考え方を学び取り入れる事は、製品品質確保につながり、リスクの低減、新しいISO13485にも準拠し、特に受託設計、製造会社 ( ODM、OEM ) では会社のプレゼンスを高め、サプライヤ−としての評価を高めます。 クオリス・イノーバのQSRコースは、多くのFDA査察準備、対応の経験や品質管理の経験から、FDA規制のみならず医療機器開発・製造の本質を鋭く説き、多くの企業の皆様の共感を集め支持されて参りました。米国に進出する企業のみならず、医療機器・IVD を開発しようとしている企業の皆様のマインドが大きく変わる価値あるクオリス・イノーバのメインコースです。
オンサイトでも可能です。お問い合わせはこちらから。
日 時:2018年10月15日(月)、16日(火) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
ODM、OEMとして設計・製造を受託している企業
受講料: ¥99,000( 2日分、テキスト・対訳本(3版)・昼食代含む )
割 引: ¥85,000
1社2名様以上ご参加の場合
特 割: 企業で行うプロジェクトの後、継続研修などでご利用の場合、
特別割引を適用させて頂きますのでメールでご相談下さい。
この場合、正規料金をお支払いの方が優先となりご希望に添えない場合が
ございますので予めご了承ください。
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
第1日目 セミナー内容
A.FDA規制、査察の最新情報と傾向分析
1.FDA規制、医療機器規制の本質( 医療機器開発・製造の本質を理解する )
2.FDA査察の本質、TQMを理解する
3.なぜISOではFDA査察にパスしないのか
4.ISOのマインドを変えないとFDA査察にパスしない
5.根拠に基づくエビデンスの重要性
6,QSITアプローチとは
7.医療機器規制を正しく理解する為に
8.QSR規制要求事項の本質を理解する
820.20 経営者の責任
経営者がリードするTQMが基本
マネジメントレビューの本質、プロセスアプローチ
プロセスを常に改善する
820.22 品質監査
効果的な監査手法と監査報告のあり方
内部監査はプロセスの異常を見極める
内部監査員はなぜ異常を見極められないのか
820.25 要員
教育の重要性:教育の手法と記録、力量表、リソースとは
教育と訓練の相違、アウェアネストレーニングの実施と訓練の重要性
820.30 設計管理( 概要のみ:11月開催 設計監理2日コースで詳細解説 )
ライフサイクル製品開発手法、ウオーターフォールモデル、
フロントローディング手法、ロバスト設計
設計検証と設計バリデーションの相違
今までISO になかった設計移管、工程設計とは
設計CAPAとは
820.40 文書管理
グロバールに通用する文書管理
品質を担保するSOPの作成方法
820.50 購買管理
開発購買とその役割、購買プロセス
3つの購買(製品、サービス、一般)と購買製品検証
新規サプライヤ評価と継続評価
購買情報とは
820.60 識別
混同を防止する為の識別方法とは
820.65 トレーサビリティー
体内埋込型機器だけではなく、変化点を知り品質を高める
820.80 アクセプタンスアクティビティー
3つのアクセプタンスを理解する
Receiving, In-process, and Finished device acceptance
820.86 アクセプタンス状態
状態表示する意味とは
820.90 不適合品
不適合品の処理プロセス
FDAはなぜNCを見たがるのか
グローバル用語を使用し、CAPAとのリンクを示す
820.120 機器のラベリング
リコールが一番多く査察で注目されるラベリングの出納管理
ラベリングの設計とは
ラベルとラベリングの相違
820.130 機器の梱包
梱包箱を見ればどんな設計を行っていたかが判る
設計依託先はISOで十分か?
820.140 取扱い
混同・劣化・損傷・汚染を発生させないためのリスク管理
820.150 保管
リスクベース保管管理
820.160 流通
ユーザーに届けるまでの製品設計
リスクベース流通管理
820.170 据付け
インスタレーション手順書のV&V
820.180 記録
全ての活動が根拠に基づくエビデンスで保証されるための重要な活動
Objective Evidence を示すための記録管理とは
820.181 機器原簿(DMR)
DMRを定義し識別する
DHF,DMR,DHRの相違
820.184 機器履歴簿(DHR)
製造記録の記載方法
コンプライアンス、製品品質を立証するため重要なDHR
820.186 品質システム記録
DHF,DMR,DHR以外の品質記録とは
820.200 付帯サービス
サービス記録の分析とMR報告,MDRの判定
820.250 統計的手法
TQMを進めていく上で最も重要な要素
品質改善に欠かせない統計的手法
第2日目 セミナー内容
820.70 生産及び工程管理
工程設計と工程V&V
( 概要のみ:11月開催 設計監理2日コースで詳細解説 )
工程のリスクマネジメント(PFMEA)
工程を監視、管理する工程パラメータとは
要員、環境、汚染管理
設備の妥当性とは
保守と検査の相違
副資材の定義と管理
820.72 検査・測定及び試験装置
意図した用途への適合性、測定器の精度管理とは
測定器の精度管理と救済処置
820.75 工程の妥当性確認
QSR,医療機器規制の本質、工程バリデーションとは
( 概要のみ:11月開催 設計監理2日コースで詳細解説 )
TQMが目指すべき試験レスとは
バリデーションの手法( VMP,DQ、IQ、OQ、PQ、PPQ )
設備バリデーションとは
CSV( コンピュータシステムバリデーション )とは
820.198 苦情ファイル
査察のキーポイント、苦情管理の目的を理解する
苦情処理とMDR( Part 803 )、CAR( Part 806 ) の関わり
( 概要のみ:MDRコースで詳細解説 )
820.100 是正処置及び予防処置( CAPA )
FDA警告書で最も多い指摘CAPAの原則を理解する
品質システムの本質、プロセスアプローチの重要な要素
修正、是正、予防の相違を正確に理解しISOマインドを変える
CAPA7ステップを理解する
CAPAの効果(再発防止)を出す仕組み
MRで監視すべきCAPAプロセス
( 概要のみ:CAPAコースで詳細解説 )
( 内容が変更になる場合もございますが大きな変更はございません。)
2018_5 QSR Seminar 様子

FDA 設計管理2日間コース Rev.20
? リスクをヘッジしコンプライアンスを堅持する仕組み ?
21 CFR Part 820.30
このコースの売上金の一部は人道活動支援に寄付されます
本年12月開催、FDA QSR内部監査員コース出席要件コースです
5月開催QSR2日間コースも出席要件となりますので併せてご出席下さい
( オンサイト、及び過去受講された方は要件を満たします )
☆ 開催概要 ☆
ほとんどの日本企業に於ける医療機器開発・設計管理の仕組みは世界標準にほど遠く、これは、グローバルに進出しようとする企業のみならず、患者の命を預かる全ての医療機器メーカーにとって大きなリスクを伴います。 米国FDAは自らの査察で医療機器品質不良の根本原因は上流側の設計管理にあると査察の結果から統計的に見抜いており、それはクオリス・イノーバの10年以上にわたるGAP監査でも明らかです。 そこで本コースでは、グローバルな開発・設計管理を米国FDA「設計管理ガイダンス」、及び欧米で取り入れられている開発フレームワークを元に体系的に解説、また、品質不良を発生させない仕組み(ロバスト設計)をタグチメソッドと設計CAPAを用いて解説します。 2日目は、プロセスバリデーションの主旨でもある工程内試験に頼らない、均一で高品質な製品を一貫して製造する為、パラメータによる製造管理の方法を実現する工程設計(設計移管)について解説します。 ”規制” だから、ではなく、設計品質を高めることにより患者さんの命を守り、企業にとっても大きな潜在的利益を得られるという大原則は、エンジニアの方々から多くの共感と支持を頂いてきたクオリス・イノーバの最も人気のある価値あるコースです。
このコースではオンサイトでも実施しています。お問い合わせはこちらから。
日 時: 2018年6月4日(月曜)、5日(火曜) 10:00?16:30
会 場: 全国町村会館(東京・永田町) 永田町駅から徒歩1分
講 師: クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象: 医療機器・IVD市場にこれから参入しようと考えている企業
医療機器・IVDを海外に輸出、輸出しようとしている企業
FDA査察準備、並びにMAPを受けようと準備している企業
既にFDA査察を終え指摘対応に苦慮している企業
受講料: ¥99,000( テキスト・昼食代含む )
割 引: ¥85,000( テキスト・昼食代含む )
1社2名以上受講される場合
5月開催QSR2日間コースを受講された方
定 員: 24名
出席者全員に教育記録として利用できる修了書をお渡しています。
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡いたします。
2名様以上ご参加の場合、お名前とメールアドレスを連絡欄にご記入下さい。
持参品: 弊社QSR翻訳本をご持参下さい。お持ちで無い方はこちらからお求め頂けます。
ご注意: 本コースは、品質システムの一部を開設するため、システム全体の知識が無いと
理解ができません。 そこで5月に開催されるQSR2日間コースの受講をお勧め
致します。( 割引適用)
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
第1日目セミナー内容
1.設計品質の本質的価値
TQMに欠かせないマネジメントチームの重要な役割
設計環境、開発リソースの見極め
要員教育の重要性( 820.25 )
力量表と教育計画
2.820.30( a ) 総則
繰り返される設計品質に起因する製品不良
ISOとQSRの本質的な相違
理解すべき規制・規格・ガイダンス
FDAが望んでいるTQMの本質、品質工学に基づくロバスト設計
医療機器の設計手法( Water fall model )
一貫性を問うFDA査察
3.820.30( b ) 設計及び開発計画
医療機器開発ライフサイクルマネジメント(LCM)
リスクマネジメントライフサイクル
タグチメソッドの開発方式によるフロントローディング手法
PM,PLの役割
カスタマーニーズを要求仕様に変換する品質機能展開
設計品質確立の重要な要素、コンカレントエンジニアリング(CE)
設計計画書とは
4.820.30( c ) 設計インプット
ロバスト設計( タグチメソッド )による未然防止設計
要求仕様書から設計仕様書への変換
曖昧なニーズのスペックへの変換
重要な設計要素トレーサビリティーマトリックス(TMX)の使用
ラベリング、梱包の設計インプット
入力のバラツキを押さえたアウトプットの確認
設計アウトプットを定義し文書類を明確にする
E-BOM / M-BOM とは
製造仕様書とは
図面発行
DMRとのトレーサビリティー
6.820.30( e ) 設計レビュー
問題の早期発見、解決が目的で、セレモニーで決して終わらない
設計審査で設計のロバストネスを確認
設計審査とテクニカルレビュー
キーとなる審査者
7.820.30( f ) 設計検証
設計検証と設計バリデーションの相違
インプットからアウトプットまでトレーサブルであるとは
エビデンスに基づく設計
設計検証計画のポイント
設計検証レポートはデータの信頼性を確保する
8.820.30( g ) 設計バリデーション
設計バリデーションの手法
社内、臨床評価の相違と記録
ソフトウェアバリデーションとVモデル
3種類のバリデーションの相違
9.820.30( h ) 設計移管 → 第2日目(工程設計)
10.820.30( i ) 設計変更
重要な査察ポイント
設計変更プロセスとリスクアセスメント
コミュニケーションエラー
製品不良を再発させない重要な要素、設計CAPAとは
文書、図面の変更管理
11.820.30( j ) 設計履歴ファイル(DHF)
DHF / DMR / DHR の定義を理解する
識別、ファイルの基本
文書体系と設計標準
第2日目セミナー内容
設計移管と工程設計
1.なぜ工程バリデーションを実施するのか
プロセスバリデーションの目的とその本質
プロセスバリデーションの効果
工程管理の目的と本質
医療機器規制・規格を理解する
820.30(h)設計移管
2.バリデーションすべき工程とは
バリデーションすべき工程の識別
検証工程とバリデーション工程の違い
設備バリデーションと工程バリデーション
バリデーション工程の例
3.バリデーションの種類
バリデーションの種類
設備バリデーションと劣化パラメータ
コンピュータシステムバリデーション(CSV)
4.工程設計とバリデーションステップ
VMP: バリデーションマスタープラン
DQ: 設計時的確性評価
IQ: 据付時的確性評価
OQ: 稼働時的確性評価
PQ: 製造性能的確性評価
PPQ: プロセス稼働性評価
バリデーションの文書化
プロセスバリデーションの明確化
自動化工程の明確化
CSVカテゴリーの明確化
ソフトウェアバリデーションの基本
カテゴリー別ソフトウェア
5.820.30( h ) 設計移管
820.75 プロセスバリデーション
820.72 検査、測定及び試験装置
設計移管とプロセスバリデーションの関係
EVとPV、検査測定器の妥当性とは
ミニスケールとフルスケール
工程設計表とトレーサビリティーマトリックス(PTMX)
6.バリデーションの実践
バリデーションマスタープラン(VMP)
バリデーション工程の識別
バリデーション計画
設備リストとバリデーション工程リスト
チームビルディング
DQ Plan /DQ Report
IQ Plan/IQ Report
OQ Plan/OQ Report
PQ Plan/PQ Report
PPQ Plan / PPQ Report
7.バリデーションのアウトプット
QC工程表と管理図
工程能力
再バリデーションの時期設定
設備メンテナンス手順
( 内容が変更にある場合もございますが、本質は変わりません )

2017_11 設計管理2日間コース
FDA QSR2日間コース Rev.20
? 医療機器・IVDに求められる規制要求事項の本質を学ぶ ?
21CFR Part 820
定員オーバーにつき募集を締め切りました
次回のご参加をお待ちしております
このコースの売り上げ金の一部は人道支援活動に寄付されます
12月開催 FDA 内部監査員コースの出席される場合の要件コースです
設計管理コースを受講される方も本コースを受講されることをお勧めします
☆ 開催概要 ☆
* FDA 品質システム規制 QSR 対訳本( 第2版 )付き
グローバルに進出しようとする企業にとって 米国は最も大きなマーケットであり、米国市場での成功がグローバルを制するといっても過言ではありません。しかし、米国に上市するには医療機器規制QSRに基づくFDAの査察は避けられません。( MDSAPを受審される企業も必須 )また、医療機器を国内で販売する際にもFDA QSRのシステムフレームワーク、エビデンスベースの考え方を学び取り入れる事は、製品品質確保につながり、リスクの低減、新しいISO13485にも準拠し、特に受託設計、製造会社 ( ODM、OEM ) では会社のプレゼンスを高め、サプライヤ−としての評価を高めます。 クオリス・イノーバのQSRコースは、多くのFDA査察準備、対応の経験や品質管理の経験から、FDA規制のみならず医療機器開発・製造の本質を鋭く説き、多くの企業の皆様の共感を集め支持されて参りました。米国に進出する企業のみならず、医療機器・IVD を開発しようとしている企業の皆様のマインドが大きく変わる価値あるクオリス・イノーバのメインコースです。
オンサイトでも可能です。お問い合わせはこちらから。
日 時:2018年5月14日(月)、15日(火) 10:00 ? 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
ODM、OEMとして設計・製造を受託している企業
受講料: ¥99,000( 2日分、テキスト・対訳本・昼食代含む )
割 引: ¥85,000( 割引適用、テキスト・対訳本・昼食代含む )
1社2名様以上ご参加の場合
特 割: 企業で行うプロジェクトの後、継続研修などでご利用の場合、
特別割引を適用させて頂きますのでメールでご相談下さい。
この場合、正規料金をお支払いの方が優先となりご希望に添えない場合が
ございますので予めご了承ください。
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
第1日目 セミナー内容
A.FDA規制、査察の最新情報と傾向分析
1.FDA規制、医療機器規制の本質( 医療機器開発・製造の本質を理解する )
2.FDA査察の本質、TQMを理解する
3.なぜISOではFDA査察にパスしないのか
4.ISOのマインドを変えないとFDA査察にパスしない
5.根拠に基づくエビデンスの重要性
6,QSITアプローチとは
7.医療機器規制を正しく理解する為に
8.QSR規制要求事項の本質を理解する
820.20 経営者の責任
経営者がリードするTQMが基本
マネジメントレビューの本質、プロセスアプローチ
プロセスを常に改善する
820.22 品質監査
効果的な監査手法と監査報告のあり方
内部監査はプロセスの異常を見極める
内部監査員はなぜ異常を見極められないのか
820.25 要員
教育の重要性:教育の手法と記録、力量表、リソースとは
教育と訓練の相違、アウェアネストレーニングの実施と訓練の重要性
820.30 設計管理( 概要のみ:6月開催 設計監理2日コースで詳細解説 )
ライフサイクル製品開発手法、ウオーターフォールモデル、
フロントローディング手法、ロバスト設計
設計検証と設計バリデーションの相違
今までISO になかった設計移管、工程設計とは
設計CAPAとは
820.40 文書管理
グロバールに通用する文書管理
品質を担保するSOPの作成方法
820.50 購買管理
開発購買とその役割、購買プロセス
3つの購買(製品、サービス、一般)と購買製品検証
新規サプライヤ評価と継続評価
購買情報とは
820.60 識別
混同を防止する為の識別方法とは
820.65 トレーサビリティー
体内埋込型機器だけではなく、変化点を知り品質を高める
820.80 アクセプタンスアクティビティー
3つのアクセプタンスを理解する
Receiving, In-process, and Finished device acceptance
820.86 アクセプタンス状態
状態表示する意味とは
820.90 不適合品
不適合品の処理プロセス
FDAはなぜNCを見たがるのか
グローバル用語を使用し、CAPAとのリンクを示す
820.120 機器のラベリング
リコールが一番多く査察で注目されるラベリングの出納管理
ラベリングの設計とは
ラベルとラベリングの相違
820.130 機器の梱包
梱包箱を見ればどんな設計を行っていたかが判る
設計依託先はISOで十分か?
820.140 取扱い
混同・劣化・損傷・汚染を発生させないためのリスク管理
820.150 保管
リスクベース保管管理
820.160 流通
ユーザーに届けるまでの製品設計
リスクベース流通管理
820.170 据付け
インスタレーション手順書のV&V
820.180 記録
全ての活動が根拠に基づくエビデンスで保証されるための重要な活動
Objective Evidence を示すための記録管理とは
820.181 機器原簿(DMR)
DMRを定義し識別する
DHF,DMR,DHRの相違
820.184 機器履歴簿(DHR)
製造記録の記載方法
コンプライアンス、製品品質を立証するため重要なDHR
820.186 品質システム記録
DHF,DMR,DHR以外の品質記録とは
820.200 付帯サービス
サービス記録の分析とMR報告,MDRの判定
820.250 統計的手法
TQMを進めていく上で最も重要な要素
品質改善に欠かせない統計的手法
第2日目 セミナー内容
820.70 生産及び工程管理
工程設計と工程V&V
( 概要のみ:6月開催 設計監理2日コースで詳細解説 )
工程のリスクマネジメント(PFMEA)
工程を監視、管理する工程パラメータとは
要員、環境、汚染管理
設備の妥当性とは
保守と検査の相違
副資材の定義と管理
820.72 検査・測定及び試験装置
意図した用途への適合性、測定器の精度管理とは
測定器の精度管理と救済処置
820.75 工程の妥当性確認
QSR,医療機器規制の本質、工程バリデーションとは
( 概要のみ:6月開催 設計監理2日コースで詳細解説 )
TQMが目指すべき試験レスとは
バリデーションの手法( VMP,DQ、IQ、OQ、PQ、PPQ )
設備バリデーションとは
CSV( コンピュータシステムバリデーション )とは
820.198 苦情ファイル
査察のキーポイント、苦情管理の目的を理解する
苦情処理とMDR( Part 803 )、CAR( Part 806 ) の関わり
( 概要のみ:MDRコースで詳細解説 )
820.100 是正処置及び予防処置( CAPA )
FDA警告書で最も多い指摘CAPAの原則を理解する
品質システムの本質、プロセスアプローチの重要な要素
修正、是正、予防の相違を正確に理解しISOマインドを変える
CAPA7ステップを理解する
CAPAの効果(再発防止)を出す仕組み
MRで監視すべきCAPAプロセス
( 概要のみ:CAPAコースで詳細解説 )
( 内容が変更になる場合もございますが大きな変更はございません。)
2017_10 QSR Seminar

〜 指摘の最も多いCAPAの正しい理解と問題の予防と再発防止の仕組み 〜
このコースの売上金の一部は人道支援に寄付されます
定員オーバーにつき募集を締め切りました
次回のご参加をお待ちしております
* コース概要 *
米国FDAの査察結果を統計的に解析していみると、CAPAが毎年ワースト3に入っている事が判ります。これは日本企業の指摘でも顕著です。FDAは自らの査察を通して企業で最も理解されていないプロセスを統計的によく理解しており、不適合の再発原因がCAPAプロセスにある事を理解しています。このことはクオリス・イノーバが実施するGAP監査でも明かで、日本のほとんどの企業がCAPAを正しく理解しておらず、トップが品質を正しくマネジメントできていません。企業の品質管理軽視の結果は昨今の事件から明らかで、医療機器の場合、命に関わる大問題です。本コースでは、品質システムの本質、プロセスアプローチを達成する為の必須ツールであるCAPAを理解し、再発防止のみならず、未然防止を理解して頂きます。 受講者と同じ目線の御社事例を使用することでより理解が高まるオンサイトセミナーが可能です。お問い合わせはこちらから。
日 時:2018年3月5日(月曜)10:00〜16:30
会 場:全国町村会館( 東京・永田町 ) ( 永田町駅から徒歩1分 )
講 師:クオリス・イノーバ株式会社代表取締役社長 木村 浩実
対 象:医療機器、IVD、医薬品等に限定せず、CAPAを用いいた品質改善を改善したい企業
受講料: ¥50,000( テキスト代・昼食含む )
割 引: ¥40,000( テキスト代・昼食含む )
1社2名様以上ご参加の場合
特 割:弊社プロジェクトを実施中の場合や、継続研修をご希望の場合、ご負担軽減のためさらに
割引をさせて頂きますのでお問い合わせください。 この場合、正規料金をお支払いのお
客様が優先され、ご出席できない場合がございますのでご了解ください。
再受講:空席がある場合、特別な料金で再受講可能ですのでお問い合わせください。
併せて受講された日もご記入ください。
定 員:20名様 2月27日締め切り
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込:セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールで連絡いたします。
複数名ご参加の場合、同様の情報をご記入下さい。 割引料金でご案内いたします。
メールでのお問い合わせ
電話でのお問い合わせ
042−856−2208
☆ セミナー内容 ☆
1.なぜFDAはCAPAに注目するのか
2.なぜ品質問題は再発するのか
3.規制・規格を正しく理解する
4.TQMの仕組みが不可欠
トップマネジメントの理解不足が品質不良を招く
5.用語の正しい理解
修正、是正、予防の相違
逸脱、不適合、逸脱許可と特別採用の相違
修正、手直し、修理の相違
6.直接原因と根本原因の相違
根本原因の改善がプロセスアプローチの本質
7.予防プロセス
品質管理の原則、予防活動の理解
8.修正、是正のプロセス
苦情処理、不適合処理、MDR、リコールプロセス
9.エスカレーション
CAPAエスカレーション
CAPAを上手に運用するための仕組み
10.CAPA7ステップ
Step 1 問題を検出し明確にする
Step 2 問題の原因を詳しく調査する
Step 3 根本原因の特定
Step 4 予防または是正する為の処置立案
処置の検証・妥当性確認
Step 5 予防または是正処置の実施
Step 6 処置の結果を文書化
Step 7 有効性の確認
11.CARの実施
12.製品開発へのCAPA
13.CAPAマネジメント
マネジメントレビューの運用方法
14.品質問題の事例に学ぶ
Warning Letter Case Study

2017_3 CAPA
FDA MDR&CAR コース Rev.3
〜 指摘の多い有害事象報告・リコール規制の理解とプロセス構築 〜
Reference 21CFR : Part 803, 806
このコースの売り上げ金の一部は人道支援活動に寄付されます
☆ 開催概要 ☆
2016年FDA査察レポートの解析からワースト4位(全体の約10%)に急浮上しているのがMDR。 日本でも警告書を受けているほとんどの企業がMDRの指摘を受けています。昨今のデータ偽装事件を受け、さらに厳しく査察されることは間違いありません。これはMDR報告の判断に迷ったら報告するという米国の文化とは異なり、日本のメーカーは情報を積極的に発信しない、隠すという古い会社の文化が根強くあることが要因の一つですが、そもそも規制を理解せず、正しい判断を行っていないことが大きな原因です。医療機器を作っているメーカーは、患者さんの命を守るという社会的な責任があります。会社を守っているようでも、結果的に会社が倒産に追い込まれることもある非常にリスクの高いプロセスであることを理解すべきです。それには規制の正しい理解、規制を守る仕組みと組織が必要です。このセミナーでは、FDAの言うCAPAサブシステム(苦情、不適合、CAPA)の各プロセスとリンクし、日本のメーカーの Warning Letter (警告書)を引き合いに正しいMDRプロセスと関連するリコールプロセスを構築することを理解します。このコースはオンサイトでも実施しています。お問い合わせはこちらから。
日 時:2018年1月22日(月曜)10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:既に医療機器・IVDを海外に輸出している企業
既にFDA査察を終え対応に困っている企業
受講料: ¥50,000-
割 引: ¥40,000-( 1社2名様以上受講される場合 )
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
1.規制を理解する
事例と統計からみる規制違反
理解すべき規制・規格・ガイダンス
eMDR規制
規制が求めるあるべき組織と力量
用語の定義を理解する
2.規制当局が考えているプロセスとは
FDAが理解しているサブシステムとは
苦情処理、CAPA、不適合処理、リコールプロセスとのリンク
社内苦情と顧客からの苦情
規制で求められる契約書
3.MDRプロセス
苦情処理プロセス
不適合処理プロセス
CAPAプロセス
MDR報告プロセス eMDR
ディシジョンツリー
4.CAR( リコール )プロセス
改修及び回収
用語の定義を理解する
報告義務のないCARとは
CARプロセス
5.警告書から理解すべき注意点とプロセスへの展開
日本の Warning Letter ( 警告書 )の事例から理解する
警告書から理解すべき注意点とプロセスへの展開
*コース内容が一部変更される場合がございますが大きな変更はございません。
FDA QSR 内部監査員コース Rev.7
〜 QSR 対応内部監査員養成のための社内資格認定2日間コース 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
募集を締め切りました!
☆ 開催概要 ☆
不適合を出さないようプロセスに内在する不適合を未然に抽出し、マネジメントレビューでトップマネジメントの指示のもとプロセス改善を常に行う。これがTQMの有るべき姿であり規制当局が求めている姿です。一方、現実はプロセスを監視し傾向管理を行っておらず、よく訓練されていないにわか監査員がISOで要求されているからといって片手間に監査を行っているだけ。これは医療機器の場合致命的で、リコールで何十億の損失を出すだけではなく人の命に関わると会社の存続まで危ぶまれます。このコースでは、ISO内部監査員の資格を持つ方々がQSRの内部監査が行えるよう社内資格を付与するための社内資格認定コースとして位置づけられます。一般に行われているISOの内部監査手法とは異なる、付加価値を生み、予防対策としての監査手法とノウハウを学びます。( このコースを受講した後、オンサイトでクオリス・イノーバによる内部監査のOJTを行うとより効果的です )
日 時:2017年12月4日(月曜)、5日(火曜) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:すでに弊社主催下記コースをオープン、オンサイト問わず受講された方限定です。
* FDA GMP QSR 2日間コース
* FDA 設計管理2日間コース
このコースはQSRの基礎知識が無いと受講できず、弊社のコースを受講されていない場合
異なる解釈により内容をよく理解出来ません。 ご理解をお願い申し上げます。
受講料: ¥99,000( テキスト、昼食代含む、内税 )
このコースには割引がございません。
定 員: 18名限定 満席になり次第締切ます
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
過去に受講された弊社QSR、設計管理各コースの受講日を記載して下さい。
オンサイトで受講された場合でも構いません。
折り返し、会場、お振込先情報をメールでご連絡致します。
![]()
お問い合わせ: お問い合わせのページからお問い合わせ下さい
お電話の方は下記セミナー事務局までお問い合わせ下さい
042−856−2208
☆ セミナー内容 ☆
1.内部監査の目的を理解する
規制・規格が求めていること
患者の命を優先する監査とは
企業の付加価値を高める為には
2.内部監査員に求められる力量
内部監査員とリーダーに求められる力量
内部監査員訓練
マネジメントチームのカウンセラー
3.プロセスアプローチ
CAPAの理解とCAPAアプローチ
プロセスのR&R( 役割と責任 )を明確にする
プロセスを常に測定し改善する
内部監査はプロセスに注目
4.監査判定基準
チェックリストと数値化
プロセス毎に数値化
FDA査察と内部監査判断基準の相違を知る
5.監査を計画する
マネジメントレビューと品質会議からの指示
リスクベース&プロセスアプローチによる監査計画
プロセス管理パラメートと傾向分析結果
是正監査と予防監査の相違
監査は短期集中型で実施
監査員レビュー会のノウハウ
監査報告書記載のポイント
6.内部監査レポート
トップマネジメントに対する報告
プロセスの不足、改善点を報告
規制に対するコンプライアンス状況の報告
トップマネジメントのアクション
7.監査の目のつけどころ( QSIT )
QSITに基づく監査手法と勘所
1.経営者による管理
2.設計管理
3.CAPA
MDR報告
CAR
MDトラッキング
4.P&PC
5.滅菌工程
+6.各プロセス
*内容は変更される場合がございます*

2016 Internal Auditors Course
FDA 設計管理2日間コース Rev.19
〜 リスクをヘッジしコンプライアンスを堅持する仕組み 〜
21CFR Part 820.30
売上金の一部は人道支援活動に寄付されます
12月開催FDA内部監査員コースの出席条件となるコースです
定員オーバーにつき募集を締め切りました!
次回のご出席をお待ちしております
☆ 開催概要 ☆
ほとんどの日本企業に於ける開発・設計管理の仕組みは、医療機器設計の世界標準と言っても過言ではない米国FDA規制に準拠していません。これはグローバルに進出しようとする企業だけではなく、患者の命を預かる全ての医療機器メーカーにとって大きなリスクを伴います。FDAは自らの査察で医療機器品質不良の根本原因は設計管理にあると統計的に見抜いており、それはクオリス・イノーバの多くのGAP監査経験でも明かです。そこで本コースでは、グローバルな開発・設計管理を「FDA設計管理ガイダンス」と、欧米で取り入れられているフレームワークを元に体系的に解説し、医療機器開発の本質である品質不良を発生させない仕組み(ロバスト設計)をタグチメソッドと設計CAPAを用いて解説します。2日目は、ほとんどのメーカーで理解されていないプロセスバリデーションを含む工程設計を、ISOで新しく設定された設計移管を絡め理解して頂きます。 ただ ” 規制だから " ではなく、設計品質を高めると企業にとっても大きな潜在的利益を得られ、エンジニアの方から多くの共感と支持を集めてきたクオリス・イノーバの最も人気のある価値あるコースです。 このコースはオンサイトでも実施しています。お問い合わせはこちらから。
日 時:2017年11月13日(月曜)、14日(火曜) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している企業
FDA査察準備を考えている企業
既にFDA査察を終え対応に困っている企業
受講料: ¥99,000( テキスト・昼食代含む )
割 引: ¥85,000( テキスト・昼食代含む )
1社2名以上受講される場合
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
ご参加なさる方は、より理解する為に事前にQSR2日間コースを受講されることをお勧め致します。QSRコースでお配りしている弊社QSR翻訳本をご持参下さい。 お持ちでない方はこちらからお求め戴けます。
![]()
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
このコースを受講される前にQSR2日コースを受講されることをお勧め致します。
弊社主催 内部監査員コースは、QSR2日間コースと設計監理2日間コースの両方の出席が要件です。
第1日目 セミナー内容
製品設計
1.設計品質の本質的価値
繰り返される設計品質に起因する製品不良
ISOとQSRの本質的な違い
理解すべき規制・規格・ガイダンス
FDAが望んでいるTQMの本質、品質工学に基づくロバスト設計
医療機器の設計手法( Water Fall Model )
一貫性を問うFDA査察
2.820.30 ( a ) 総則
TQMに欠かせないマネジメントチームの重要な役割
設計環境、開発リソースの見極め
要員教育の重要性( 820.25 )
力量表と教育計画
3.820.30 ( b ) 設計及び開発計画
医療機器開発ライフサイクルマネジメント
リスクマネジメントライフサイクル
タグチメソッドの開発方式によるフロントローディング手法
PM,PLの役割
カスタマーニーズを要求仕様に変換する品質機能展開
設計品質確立の重要な要素、コンカレントエンジニアリング
設計計画書とは
4.820.30 ( c ) 設計インプット
ロバスト設計( タグチメソッド )による未然防止設計
要求仕様書から設計仕様書への変換
曖昧なニーズのスペックへの変換
重要な設計要素トレーサビリティーマトリックスの使用
ラベリング、梱包の設計インプット
5.820.30 ( d ) 設計アウトプット
入力のバラツキを押さえたアウトプットの確認
設計アウトプットを定義し文書類を明確にする
E-BOM / M-BOM とは
製造仕様書とは
図面発行
DMRとのトレーサビリティー
6.820.30 ( j ) 設計履歴ファイル( DHF )
DHF / DMR / DHR の定義を理解する
識別、ファイルの基本
文書体系と設計標準
7.820.30 ( e ) 設計審査
問題の早期発見、解決が目的で、セレモニーで決して終わらない
設計審査で設計のロバストネスを確認
設計審査とテクニカルレビュー
キーとなる審査者
8.820.30 ( f ) 設計検証
設計検証と設計バリデーションの相違
インプットからアウトプットまでトレーサブルであるとは
エビデンスに基づく設計
設計検証計画のポイント
設計検証レポートはデータの信頼性を確保する
9.820.30 ( g ) 設計バリデーション
設計バリデーションの手法
社内、臨床評価の相違と記録
ソフトウェアバリデーションとVモデル
3種類のバリデーションの相違
10.820.30 ( i ) 設計変更
重要な査察ポイント
設計変更プロセスとリスクアセスメント
コミュニケーションエラー
製品不良を再発させない重要な要素、設計CAPAとは
文書、図面の変更管理
第2日目 セミナー内容
工程設計
1.なぜ工程バリデーションを実施するのか?
プロセスバリデーションの目的とその本質
プロセスバリデーションの効果
工程管理の目的と本質
医療機器規制・規格を理解する
820.30(h)設計移管
2.バリデーションすべき工程とは
バリデーションすべき工程の識別
検証工程とバリデーション工程の違い
設備バリデーションと工程バリデーション
バリデーション工程の例
3.バリデーションの種類
バリデーションの種類
設備バリデーションと劣化パラメータ
コンピュータシステムバリデーション(CSV)
4.工程設計とバリデーションステップ
VMP: バリデーションマスタープラン
DQ: 設計時的確性評価
IQ: 据付時的確性評価
OQ: 稼働時的確性評価
PQ: 製造性能的確性評価
PPQ: プロセス稼働性評価
バリデーションの文書化
プロセスバリデーションの明確化
自動化工程の明確化
CSVカテゴリーの明確化
ソフトウェアバリデーションの基本
カテゴリー別ソフトウェア
5.820.30 ( h ) 設計移管と
820.75 プロセスバリデーション
820.72 検査、測定及び試験装置
設計移管とプロセスバリデーションの関係
EVとPV、検査測定器の妥当性とは
ミニスケールとフルスケール
工程設計表とトレーサビリティー
6.バリデーションの実践
マスターバリデーションプラン
バリデーション工程の識別
バリデーション計画
設備リストとバリデーション工程リスト
チームビルディング
DQ Plan /DQ Report
IQ Plan/IQ Report
OQ Plan/OQ Report
PQ Plan/PQ Report
PPQ Plan / PPQ Report
7.バリデーションのアウトプット
QC工程表と管理図
工程能力
再バリデーションの時期設定
設備メンテナンス手順
( 内容が変更になる場合もございますが大きな変更はございません。 )

2017_5 設計管理コース
FDA QSR2日間コース Rev.19
〜 医療機器・IVDに求められる規制要求事項の本質を学ぶ 〜
21CFR Part 820
このコースの売り上げ金の一部は人道支援活動に寄付されます
12月開催 FDA 内部監査員コースの出席される場合の要件コースです
設計管理コースを受講されるかも本コースを受講されることをお勧めします
定員オーバーにつき、募集を終了いたしました!
次回、皆様のご参加をお待ちしております。
☆ 開催概要 ☆
* FDA 品質システム規制 QSR 対訳本( 第2版 )付き
グローバルに進出しようとする企業にとって 米国は最も大きなマーケットであり、米国市場での成功がグローバルを制するといっても過言ではありません。しかし、米国に上市するには医療機器規制QSRに基づくFDAの査察は避けられません。( MDSAPを受審される企業も必須 )また、医療機器を国内で販売する際にもFDA QSRのシステムフレームワーク、エビデンスベースの考え方を学び取り入れる事は、製品品質確保につながり、リスクの低減、新しいISO13485にも準拠し、特に受託設計、製造会社 ( ODM、OEM ) では会社のプレゼンスを高め、サプライヤ−としての評価を高めます。 クオリス・イノーバのQSRコースは、多くのFDA査察準備、対応の経験や品質管理の経験から、FDA規制のみならず医療機器開発・製造の本質を鋭く説き、多くの企業の皆様の共感を集め支持されて参りました。米国に進出する企業のみならず、医療機器・IVD を開発しようとしている企業の皆様のマインドが大きく変わる価値あるクオリス・イノーバのメインコースです。
オンサイトでも可能です。お問い合わせはこちらから。
日 時:2017年10月16日(月)、17日(火) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
ODM、OEMとして設計・製造を受託している企業
受講料: ¥99,000( 2日分、テキスト・対訳本・昼食代含む )
割 引: ¥85,000( 割引適用、テキスト・対訳本・昼食代含む )
1社2名様以上ご参加の場合
特 割: 企業で行うオンサイトセミナー開催の後、継続研修などでご利用の場合、
特別割引を適用させて頂きますのでメールでご相談下さい。
この場合、正規料金をお支払いの方が優先となりご希望に添えない場合が
ございますので予めご了承ください。
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
第1日目 セミナー内容
A.FDA規制、査察の最新情報と傾向分析
1.FDA規制、医療機器規制の本質( 医療機器開発・製造の本質を理解する )
2.FDA査察の本質、TQMを理解する
3.なぜISOではFDA査察にパスしないのか
4.ISOのマインドを変えないとFDA査察にパスしない
5.根拠に基づくエビデンスの重要性
6,QSITアプローチとは
7.医療機器規制を正しく理解する為に
8.QSR規制要求事項の本質を理解する
820.20 経営者の責任
経営者がリードするTQMが基本
マネジメントレビューの本質、プロセスアプローチ
プロセスを常に改善する
820.22 品質監査
効果的な監査手法と監査報告のあり方
内部監査はプロセスの異常を見極める
内部監査員はなぜ異常を見極められないのか
820.25 要員
教育の重要性:教育の手法と記録、力量表、リソースとは
教育と訓練の相違、アウェアネストレーニングの実施
820.30 設計管理( 概要のみ:設計監理1日コースで詳細解説 )
ライフサイクル製品開発手法、ウオーターフォールモデル、
フロントローディング手法、ロバスト設計
設計検証と設計バリデーションの相違
今までISO になかった設計移管、工程設計とは
設計CAPAとは
820.40 文書管理
グロバールに通用する文書管理
品質に影響するSOPの作成方法
820.50 購買管理
これからの査察の潮流、サプライヤー管理
開発購買とその役割
820.60 識別
混同を防止する為の識別方法とは
820.65 トレーサビリティー
体内埋込型機器だけではなく、変化点を知り品質を高める
820.80 アクセプタンスアクティビティー
3つのアクセプタンスを理解する
Receiving, In-process, and Finished device acceptance
820.86 アクセプタンス状態
状態表示する意味とは
820.90 不適合品
不適合品の処理プロセス
FDAはなぜNCを見たがるのか
グローバル用語を使用し、CAPAとのリンクを示す
820.120 機器のラベリング
リコールが一番多く査察で注目されるラベリングの出納管理
ラベリングの設計とは
ラベルとラベリングの相違
820.130 機器の梱包
梱包箱を見ればどんな設計を行っていたかが判る
設計依託先はISOで十分か?
820.140 取扱い
混同・劣化・損傷・汚染を発生させないためのリスク管理
820.150 保管
リスクベース保管管理
820.160 流通
ユーザーに届けるまでの製品設計
リスクベース流通管理
820.170 据付け
インスタレーション手順書のV&V
820.180 記録
全ての活動が根拠に基づくエビデンスで保証されるための重要な活動
Objective Evidence を示すための記録管理とは
820.181 機器原簿(DMR)
DMRを定義し識別する
DHF,DMR,DHRの相違
820.184 機器履歴簿(DHR)
製造記録の記載方法
コンプライアンス、製品品質を立証するため重要なDHR
820.186 品質システム記録
DHF,DMR,DHR以外の品質記録とは
820.200 付帯サービス
サービス記録の分析とMR報告,MDRの判定
820.250 統計的手法
TQMを進めていく上で最も重要な要素
品質改善に欠かせない統計的手法
第2日目 セミナー内容
820.70 生産及び工程管理
工程設計と工程V&V
工程のリスクマネジメント(PFMEA)
工程を監視、管理する工程パラメータとは
要員、環境、汚染管理
設備の妥当性とは
保守と検査の相違
副資材の定義と管理
820.72 検査・測定及び試験装置
意図した用途への適合性、測定器の精度管理とは
測定器の精度管理と救済処置
820.75 工程の妥当性確認
QSR,医療機器規制の本質、工程バリデーションとは
TQMが目指すべき試験レスとは
バリデーションの手法( VMP,DQ、IQ、OQ、PQ、PPQ )
設備バリデーションとは
CSV( コンピュータシステムバリデーション )とは
820.198 苦情ファイル
査察のキーポイント、苦情管理の目的を理解する
苦情処理とMDR( Part 803 )、CAR( Part 806 ) の関わり
820.100 是正処置及び予防処置( CAPA )
FDA警告書で最も多い指摘CAPAの原則を理解する
品質システムの本質、プロセスアプローチの重要な要素
修正、是正、予防の相違を正確に理解しISOマインドを変える
CAPA7ステップを理解する
CAPAの効果(再発防止)を出す仕組み
MRで監視すべきCAPAプロセス
( 内容が変更になる場合もございますが大きな変更はございません。)
2017_5 QSR Seminar

FDA設計管理2日間コース Rev.18
〜 規制だけではなく会社に利益をもたらすものづくりの本質 〜
Reference 21CFR: 820 Subpart C, G
このコースの売り上げ金の一部は人道支援活動に寄付されます
12月開催 FDA 内部監査員コースの出席条件となるコースです
終了しました!
☆ 開催概要 ☆
ほとんどの日本企業に於ける開発・設計管理の仕組みは、医療機器設計の世界標準と言っても過言ではない米国FDA規制に準拠していません。これはグローバルに進出しようとする企業だけではなく、患者の命を預かる全ての医療機器メーカーにとって大きなリスクを伴います。FDAは自らの査察で医療機器品質不良の根本原因は設計管理にあると統計的に見抜いており、それはクオリス・イノーバの多くのGAP監査経験でも明かです。そこで本コースでは、グローバルな開発・設計管理を「FDA設計管理ガイダンス」と、欧米で取り入れられているフレームワークを元に体系的に解説し、医療機器開発の本質である品質不良を発生させない仕組み(ロバスト設計)をタグチメソッドと設計CAPAを用いて解説します。2日目は、ほとんどのメーカーで理解されていないプロセスバリデーションを含む工程設計を、ISOで新しく設定された設計移管を絡め理解して頂きます。 ただ ” 規制だから " ではなく、設計品質を高めると企業にとっても大きな潜在的利益を得られ、エンジニアの方から多くの共感と支持を集めてきたクオリス・イノーバの最も人気のある価値あるコースです。 このコースはオンサイトでも実施しています。お問い合わせはこちらから。
日 時:2017年5月29日(月曜)、30日(火曜) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している企業
FDA査察準備を考えている企業
既にFDA査察を終え対応に困っている企業
受講料: ¥99,000( テキスト・昼食代含む )
割 引: ¥85,000( テキスト・昼食代含む )
1社2名以上受講される場合
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
ご参加なさる方は、より理解する為に事前にQSR2日間コースを受講されることをお勧め致します。
QSRコースでお配りしている弊社QSR翻訳本をご持参下さい。 お持ちでない方はこちらから
お求め戴けます。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
このコースを受講される前にQSR2日コースを受講されることをお勧め致します。
弊社主催 内部監査員コースは、QSR2日間コースと設計監理2日間コースの両方の出席が要件です。
第1日目 セミナー内容
1.設計品質の本質的価値
繰り返される設計品質に起因する製品不良
ISOとQSRの本質的な違い
理解すべき規制・規格・ガイダンス
FDAが望んでいるTQMの本質、品質工学に基づくロバスト設計
医療機器の設計手法( Water Fall Model )
一貫性を問うFDA査察
〜 TQM、FDA設計管理ガイダンスに基づくQSRサブパートCの実践 〜
2.820.30 ( a ) 総則
TQMに欠かせないマネジメントチームの重要な役割
設計環境、開発リソースの見極め
要員教育の重要性( 820.25 )
力量表と教育計画
3.820.30 ( b ) 設計及び開発計画
医療機器開発ライフサイクルマネジメント
リスクマネジメントライフサイクル
タグチメソッドの開発方式によるフロントローディング手法
PM,PLの役割
カスタマーニーズを要求仕様に変換する品質機能展開
設計品質確立の重要な要素、コンカレントエンジニアリング
設計計画書とは
4.820.30 ( c ) 設計インプット
ロバスト設計( タグチメソッド )による未然防止設計
要求仕様書から設計仕様書への変換
曖昧なニーズのスペックへの変換
重要な設計要素トレーサビリティーマトリックスの使用
ラベリング、梱包の設計インプット
5.820.30 ( d ) 設計アウトプット
入力のバラツキを押さえたアウトプットの確認
設計アウトプットを定義し文書類を明確にする
E-BOM / M-BOM とは
製造仕様書とは
図面発行
DMRとのトレーサビリティー
6.820.30 ( j ) 設計履歴ファイル( DHF )
DHF / DMR / DHR の定義を理解する
識別、ファイルの基本
文書体系と設計標準
7.820.30 ( e ) 設計審査
問題の早期発見、解決が目的で、セレモニーで決して終わらない
設計審査で設計のロバストネスを確認
設計審査とテクニカルレビュー
キーとなる審査者
8.820.30 ( f ) 設計検証
設計検証と設計バリデーションの相違
インプットからアウトプットまでトレーサブルであるとは
エビデンスに基づく設計
設計検証計画のポイント
設計検証レポートはデータの信頼性を確保する
9.820.30 ( g ) 設計バリデーション
設計バリデーションの手法
社内、臨床評価の相違と記録
ソフトウェアバリデーションとVモデル
3種類のバリデーションの相違
10.820.30 ( i ) 設計変更
重要な査察ポイント
設計変更プロセスとリスクアセスメント
コミュニケーションエラー
製品不良を再発させない重要な要素、設計CAPAとは
文書、図面の変更管理
第2日目 セミナー内容
1.なぜ工程バリデーションを実施するのか?
プロセスバリデーションの目的とその本質
プロセスバリデーションの効果
工程管理の目的と本質
医療機器規制・規格を理解する
820.30(h)設計移管
2.バリデーションすべき工程とは
バリデーションすべき工程の識別
検証工程とバリデーション工程の違い
設備バリデーションと工程バリデーション
バリデーション工程の例
3.バリデーションの種類
バリデーションの種類
設備バリデーションと劣化パラメータ
コンピュータシステムバリデーション(CSV)
4.工程設計とバリデーションステップ
VMP: バリデーションマスタープラン
DQ: 設計時的確性評価
IQ: 据付時的確性評価
OQ: 稼働時的確性評価
PQ: 製造性能的確性評価
PPQ: プロセス稼働性評価
バリデーションの文書化
プロセスバリデーションの明確化
自動化工程の明確化
CSVカテゴリーの明確化
ソフトウェアバリデーションの基本
カテゴリー別ソフトウェア
5.820.30 ( h ) 設計移管と
820.75 プロセスバリデーション
820.72 検査、測定及び試験装置
設計移管とプロセスバリデーションの関係
EVとPV、検査測定器の妥当性とは
ミニスケールとフルスケール
工程設計表とトレーサビリティー
6.バリデーションの実践
マスターバリデーションプラン
バリデーション工程の識別
バリデーション計画
設備リストとバリデーション工程リスト
チームビルディング
DQ Plan /DQ Report
IQ Plan/IQ Report
OQ Plan/OQ Report
PQ Plan/PQ Report
PPQ Plan / PPQ Report
7.バリデーションのアウトプット
QC工程表と管理図
工程能力
再バリデーションの時期設定
設備メンテナンス手順
( 内容が変更になる場合もございますが大きな変更はございません。 )
 2016_11
2016_11
FDA QSR 2日間コース Rev.18
〜 医療機器・IVDに求められる規制要求事項の本質を学ぶ実践コース 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
12月開催 FDA 内部監査員コースに出席される場合の要件コースです
設計管理コースを受講される方もこのコースを受講されることをお勧め致します
☆ 開催概要 ☆
* FDA 品質システム規制 QSR 対訳本( 第2版 )付き
グローバルに進出しようとする企業にとって 米国は最も大きなマーケットであり、米国市場での成功がグローバルを制するといっても過言ではありません。しかし、米国に上市するには医療機器規制QSRに基づくFDAの査察は避けられません。( MDSAPを受審される企業も必須 )また、医療機器を国内で販売する際にもFDA QSRのシステムフレームワーク、エビデンスベースの考え方を学び取り入れる事は、リスクの低減、新しいISO13485にも準拠し、特に受託設計、製造会社 ( OEM ) では会社のプレゼンスを高め、サプライヤ−としての評価を高めます。 クオリス・イノーバのQSRコースは、多くのFDA査察準備、対応の経験や品質管理の経験から、FDA規制のみならず医療機器開発・製造の本質を鋭く説き、多くの企業の皆様の共感を集め支持されて参りました。米国に進出する企業のみならず、医療機器・IVD を開発しようとしている企業の皆様のマインドが大きく変わる価値あるクオリス・イノーバのメインコースです。
オンサイトでも可能です。お問い合わせはこちらから。
日 時:2017年5月15日(月)、16日(火) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
OEMとして設計・製造を受託している企業
受講料: ¥99,000( 2日分、テキスト・対訳本・昼食代含む )
割 引: ¥85,000( 割引適用、テキスト・対訳本・昼食代含む )
1社2名様以上ご参加の場合
特 割: 企業で行うオンサイトセミナー開催の後、継続研修などでご利用の場合、
特別割引を適用させて頂きますのでメールでご相談下さい。
この場合、正規料金をお支払いの方が優先となりご希望に添えない場合が
ございますので予めご了承ください。
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
第1日目 セミナー内容
A.FDA規制、査察の最新情報と傾向分析
1.FDA規制、医療機器規制の本質( 医療機器開発・製造の本質を理解する )
2.FDA査察の本質、TQMを理解する
3.なぜISOではFDA査察にパスしないのか
4.ISOマインドを変える
5.根拠に基づくエビデンスの重要性
6,QSITアプローチとは
7.医療機器規制を正しく理解する為に
8.QSR規制要求事項の本質を理解する
820.20 経営者の責任
経営者がリードするTQMを理解すると見えてくる
マネジメントレビューの本質 プロセスアプローチ
品質を改善しないリスク
820.22 品質監査
効果的な監査手法と監査報告のあり方
内部監査でなぜプロセス異常を見つけられないのか
820.25 要員
教育の重要性:教育の手法と記録、力量表、リソースとは
アウェアネストレーニングの実施
820.30 設計管理( 概要のみ:設計監理1日コースで詳細解説 )
ライフサイクル製品開発手法、ウオーターフォールモデル、
フロントローディング手法、ロバスト設計
設計検証と設計バリデーションの相違
今までISO になかった設計移管、工程設計とは
設計CAPAとは
820.40 文書管理
グロバールに通用する文書管理手法と教育
820.50 購買管理
これからの査察の潮流、サプライヤー管理とは
購買チームが品質に貢献するために
820.60 識別
混同を防止する為の識別方法とは
820.65 トレーサビリティー
体内埋込型機器だけではなく、変化点を知り品質を高める
820.80 アクセプタンスアクティビティー
3つのアクセプタンスを理解する
Receiving, In-process, and Finished device acceptance
820.86 アクセプタンス状態
状態表示する意味とは
820.90 不適合品
不適合品の処理プロセス
FDAはなぜNCを見たがるのか
グローバル用語を使用し、CAPAとのリンクを示す
820.120 機器のラベリング
リコールが一番多く査察で注目されるラベリングの出納管理
ラベリングの設計とは
ラベルとラベリングの相違
820.130 機器の梱包
梱包箱を見ればどんな設計を行っていたかが判る
設計依託先はISOで十分か?
820.140 取扱い
混同・劣化・損傷・汚染を発生させないためのリスク管理
820.150 保管
倉庫などの保管エリアのリスク管理
リスクアセスメントをすると。。
820.160 流通
劣化品の流通を防ぐ設計品質とは
温度指定製品の流通保証とは
820.170 据付け
インスタレーション手順書のV&V
820.180 記録
全ての活動が根拠に基づくエビデンスで保証されるための重要な活動
Objective Evidence を示すための記録管理とは
エビデンスベースの基礎を学ぶ
820.181 機器原簿(DMR)
DMRを定義し識別する
DHF,DMR,DHRの相違
820.184 機器履歴簿(DHR)
製造記録の記載方法
コンプライアンス上、特に重要なDHR
820.186 品質システム記録
DHF,DMR,DHR以外の品質記録とは
820.200 付帯サービス
サービス記録の分析とMR,MDRの判定
820.250 統計的手法
TQMを進めていく上で最も重要な要素
品質改善に欠かせない統計的手法
第2日目 セミナー内容
820.70 生産及び工程管理
工程設計と工程V&V
工程のリスクマネジメント(PFMEA)
工程を監視、管理する工程パラメータとは
要員、環境、汚染管理
設備の妥当性とは
保守と検査の相違
副資材の定義と管理
820.72 検査・測定及び試験装置
意図した用途への適合性、測定器の精度管理とは
測定器の精度管理と救済処置
820.75 工程の妥当性確認
QSR,医療機器規制の本質、工程バリデーションとは
TQMが目指すべき試験レスとは
バリデーションの手法( VMP,DQ、IQ、OQ、PQ、PPQ )
設備バリデーションとは
CSV( コンピュータシステムバリデーション )とは
820.198 苦情ファイル
査察のキーポイント、苦情管理の目的を理解する
苦情処理とMDR( Part 803 )、CAR( Part 806 )
820.100 是正処置及び予防処置( CAPA )
FDA警告書で最も多い指摘CAPAの原則を理解する
修正、是正、予防の相違を正確に理解しISOマインドを変える
CAPA7ステップを理解する
CAPAの効果(再発防止)を出す仕組み
MRで監視すべきCAPAプロセス
( 内容が変更になる場合もございますが大きな変更はございません。)
2016_11 QSR Seminar

CAPA コース Rev.16
〜 査察で最も指摘の多いCAPAの正しい理解と問題を再発させない仕組み 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
定員オーバーのため募集を終了いたしました
次回のご出席をお待ちしております
☆ 開催概要 ☆
FDA査察の結果を統計的にまとめてみるとCAPAで最も多くの指摘をしていることが判ります。これは日本企業の指摘でも顕著です。FDAは自らの査察を通して企業で最も理解されていないプロセスを統計的によく理解しており、不適合の再発原因がCAPAプロセスにあることを知っています。このことはクオリス・イノーバが実施してきたGAP監査でも明かで、日本のほとんどの企業でCAPAを正しく理解しておらずマネジメントできていません。これは品質管理が行われていないことを如実に示しており、医療の現場では非常にリスクです。また、FDAの査察を受けた後のアクションレターもCAPAのステップで回答すべきです。 このコースは参加指されたほとんどの方のCAPAのマインドが変わる価値あるコースで、医療機器だけではなく医薬品や多くの業種で理解すべき内容です。 品質システムの本質、プロセスアプローチを達成する為の必須のツールであるCAPAを理解し、再発防止のみならず、品質改善の本質、未然防止を理解して頂きます。
日 時:2017年3月6日(月曜) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVDに限らず医薬品などCAPAを用いた品質改善を期待される企業
受講料: ¥50,000( テキスト・昼食代含む )
割 引: ¥40,000( テキスト・昼食代含む )
1社2名様以上ご参加の場合
特 割: プロジェクトで実施するオンサイトセミナーの後、継続研修などでご利用戴く場合、
特別割引をさせて戴きますのでお問い合わせ下さい。 この場合、正規料金をお支払
いのお客様が優先され御出席できない場合がございますので予めご了解下さい。
再受講: 席に空きがある場合、特別料金で受講出来ます。この機会にご参加なさり、
うまくいかない点を解決しプロセスの理解を深めて下さい。
お申込の際、再受講とご記入され、併せて受講された日もご記入下さい。
定 員: 20名様 2月28日締切
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
![]()
☆ セミナー内容 ☆
1.なぜFDAはCAPAに注目するのか
2.なぜ品質問題は再発するのか
3.規制・規格を理解する
FDA 21CFR Part 820.100 CAPA vs. ISO
4.TQMの仕組みが不可欠
マネジメントチームの理解不足が品質不良を招く
5.用語を正しく理解する
修正、是正、予防の相違
不適合、逸脱、特別採用と逸脱許可の相違
修正、手直し、修理の相違
6.直接原因と根本原因を区別する
この違いの理解が最も重要
プロセスアプローチとは
7.修正、是正のプロセス
苦情処理、不適合処理プロセスとの関係
MDR,リコールプロセスとの関係
8.予防プロセス
品質管理の原則は予防活動
9.CAPAエスカレーションプロセス
CAPAプロセスをうまくまわす為に
10.CAPA7ステップ
Six Sigma との相違
Step 1 問題を検出し明確にする
Step 2 問題の原因を詳しく調査する
Step 3 根本原因を特定する
Step 4 是正又は予防するための処置立案と
処置の検証・妥当性確認
Step 5 是正または予防処置の実施
Step 6 処置の結果を文書化
Step 7 有効性の確認
11.CR/CARの実施
12.製品開発にCAPAを活かす
13.CAPAのマネジメント
品質システム履行に欠かせないプロセスアプローチの理解
マネジメントレビューの運用方法
14.品質問題の事例に学ぶ
Warning Letter Case Study
実例に学ぶ
* 内容が一部変更となる場合がございます

FDA QSR 内部監査員コース Rev.6
〜 QSR 対応内部監査員養成のための社内資格認定2日間コース 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
終了しました
☆ 開催概要 ☆
不適合を出さないようプロセスに内在する不適合を未然に抽出し、マネジメントレビューでトップマネジメントの指示のもとプロセス改善を常に行う。これがTQMの有るべき姿であり規制当局が求めている姿です。一方、現実はプロセスを監視し傾向管理を行っておらず、よく訓練されていないにわか監査員がISOで要求されているからといって片手間に監査を行っているだけ。これは医療機器の場合致命的で、リコールで何十億の損失を出すだけではなく人の命に関わると会社の存続まで危ぶまれます。このコースでは、ISO内部監査員の資格を持つ方々がQSRの内部監査が行えるよう社内資格を付与するための社内資格認定コースとして位置づけられます。一般に行われているISOの内部監査手法とは異なる、付加価値を生み、予防対策としての監査手法とノウハウを学びます。( このコースを受講した後、オンサイトでクオリス・イノーバによる内部監査のOJTを行うとより効果的です )
日 時:2016年12月5日(月曜)、6日(火曜) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:すでに弊社主催下記コースをオープン、オンサイト問わず受講された方限定です。
* FDA GMP QSR 2日間コース
* FDA 設計管理2日間コース
このコースはQSRの基礎知識が無いと受講できず、弊社のコースを受講されていない場合
異なる解釈により内容をよく理解出来ません。 皆様には何卒ご理解をお願い申し上げます。
受講料: ¥99,000( テキスト、昼食代含む、内税 )
このコースには割引がございません。
定 員: 18名限定 満席になり次第締切ます
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
過去に受講された弊社QSR、設計管理各コースの受講日を記載して下さい。
オンサイトで受講された場合でも構いません。
折り返し、会場、お振込先情報をメールでご連絡致します。
![]()
お問い合わせ: お問い合わせのページからお問い合わせ下さい
お電話の方は下記セミナー事務局までお問い合わせ下さい
042−856−2208
☆ セミナー内容 ☆
1.内部監査の目的を理解する
規制・規格が求めるていること
患者の命を優先する監査とは
企業の付加価値を高める為には
2.内部監査員に求められる力量
内部監査員とリーダーに求められる力量
内部監査員トレーニング
マネジメントチームのカウンセラー
3.プロセスアプローチ
CAPAの理解とCAPAアプローチ
プロセスのR&Rを明確にする
プロセスを常に測定し改善する
内部監査はプロセスに注目
4.監査判定基準
チェックリストと数値化
プロセス毎に数値化
FDA査察と内部監査判断基準の相違を知る
5.監査を計画する
マネジメントレビューと品質会議
プロセス管理パラメートと傾向分析結果
是正監査と予防監査の相違
監査は短期集中型で実施
監査員レビュー会のノウハウ
監査報告書記載のポイント
6.監査の目のつけどころ( QSIT )
QSITに基づく監査手法と勘所
1.経営者による管理
2.設計管理
3.CAPA
MDR報告
CAR
MDトラッキング
4.P&PC
5.滅菌工程
*内容は変更される場合がございます*
前回講習風景 2016_7

FDA設計管理2日間コース Rev.17
〜 規制だけではなく会社に利益をもたらすものづくりの本質 〜
Reference 21CFR: 820 Subpart C, G
このコースの売り上げ金の一部は人道支援活動に寄付されます
12月開催 FDA 内部監査員コースの出席条件となるコースです
☆ 開催概要 ☆
ほとんどの日本企業に於ける開発・設計管理の仕組みは、医療機器設計の世界標準と言っても過言ではない米国FDA規制に準拠していません。これはグローバルに進出しようとする企業だけではなく、患者の命を預かる全ての医療機器メーカーにとって大きなリスクを伴います。FDAは自らの査察で医療機器品質不良の根本原因は設計管理にあると統計的に見抜いており、それはクオリス・イノーバの多くのGAP監査経験でも明かです。そこで本コースでは、グローバルな開発・設計管理を「FDA設計管理ガイダンス」と、欧米で取り入れられているフレームワークを元に体系的に解説し、医療機器開発の本質である品質不良を発生させない仕組み(ロバスト設計)をタグチメソッドと設計CAPAを用いて解説します。ただ ” 規制だから " ではなく、設計品質を高めると企業にとっても大きな潜在的利益を得られ、エンジニアの方から多くの共感と支持を集めてきたクオリス・イノーバの最も人気のある価値あるコースです。 このコースはオンサイトでも実施しています。お問い合わせはこちらから。
日 時:2016年11月29日(火曜)、30日(水曜) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している企業
FDA査察準備を考えている企業
既にFDA査察を終え対応に困っている企業
受講料: ¥99,000( テキスト・昼食代含む )
割 引: ¥85,000( テキスト・昼食代含む )
1社2名以上受講される場合
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
ご参加なさる方は、より理解する為に事前にQSR2日間コースを受講されることをお勧め致します。
QSRコースでお配りしている弊社QSR翻訳本をご持参下さい。 お持ちでない方はこちらから
お求め戴けます。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
このコースを受講される前にQSR2日コースを受講されることをお勧め致します。
弊社主催 内部監査員コースは、QSR2日間コースと設計監理2日間コースの両方の出席が要件です。
第1日目 セミナー内容
1.設計品質の本質的価値
繰り返される設計品質に起因する製品不良
ISOとQSRの本質的な違い
理解すべき規制・規格・ガイダンス
FDAが望んでいるTQMの本質、品質工学に基づくロバスト設計
医療機器の設計手法( Water Fall Model )
一貫性を問うFDA査察
〜 TQM、FDA設計管理ガイダンスに基づくQSRサブパートCの実践 〜
2.820.30 ( a ) 総則
TQMに欠かせないマネジメントチームの重要な役割
設計環境、開発リソースの見極め
要員教育の重要性( 820.25 )
力量表と教育計画
3.820.30 ( b ) 設計及び開発計画
医療機器開発ライフサイクルマネジメント
リスクマネジメントライフサイクル
タグチメソッドの開発方式によるフロントローディング手法
PM,PLの役割
カスタマーニーズを要求仕様に変換する品質機能展開
設計品質確立の重要な要素、コンカレントエンジニアリング
設計計画書とは
4.820.30 ( c ) 設計インプット
ロバスト設計( タグチメソッド )による未然防止設計
要求仕様書から設計仕様書への変換
曖昧なニーズのスペックへの変換
重要な設計要素トレーサビリティーマトリックスの使用
ラベリング、梱包の設計インプット
5.820.30 ( d ) 設計アウトプット
入力のバラツキを押さえたアウトプットの確認
設計アウトプットを定義し文書類を明確にする
E-BOM / M-BOM とは
製造仕様書とは
図面発行
DMRとのトレーサビリティー
6.820.30 ( j ) 設計履歴ファイル( DHF )
DHF / DMR / DHR の定義を理解する
識別、ファイルの基本
文書体系と設計標準
7.820.30 ( e ) 設計審査
問題の早期発見、解決が目的で、セレモニーで決して終わらない
設計審査で設計のロバストネスを確認
設計審査とテクニカルレビュー
キーとなる審査者
8.820.30 ( f ) 設計検証
設計検証と設計バリデーションの相違
インプットからアウトプットまでトレーサブルであるとは
エビデンスに基づく設計
設計検証レポートは、設計者の活動記録
9.820.30 ( g ) 設計バリデーション
設計バリデーションの手法
社内、臨床評価の相違と記録
ソフトウェアバリデーションとVモデル
3種類のバリデーションの相違
10.820.30 ( i ) 設計変更
重要な査察ポイント
設計変更プロセスとリスクアセスメント
コミュニケーションエラー
製品不良を再発させない重要な要素、設計CAPAとは
文書、図面の変更管理
第2日目 セミナー内容
1.なぜ工程バリデーションを実施するのか?
プロセスバリデーションの目的とその本質
プロセスバリデーションの効果
工程管理の目的と本質
医療機器規制・規格を理解する
820.30(h)設計移管
2.バリデーションすべき工程とは
バリデーションすべき工程の識別
検証工程とバリデーション工程の違い
設備バリデーションと工程バリデーション
バリデーション工程の例
3.バリデーションの種類
バリデーションの種類
設備バリデーションと劣化パラメータ
コンピュータシステムバリデーション(CSV)
4.工程設計とバリデーションステップ
VMP: バリデーションマスタープラン
DQ: 設計時的確性評価
IQ: 据付時的確性評価
OQ: 稼働時的確性評価
PQ: 製造性能的確性評価
PPQ
バリデーションの文書化
プロセスバリデーションの明確化
自動化工程の明確化
CSVカテゴリーの明確化
ソフトウェアバリデーションの基本
カテゴリー別ソフトウェア
5.820.30 ( h ) 設計移管と
820.75 プロセスバリデーション
820.72 検査、測定及び試験装置
設計移管とプロセスバリデーションの関係
EVとPV、検査測定器の妥当性とは
ミニスケールとフルスケール
工程設計表とトレーサビリティー
6.バリデーションの実践
マスターバリデーションプラン
バリデーション工程の識別
バリデーション計画
設備リストとバリデーション工程リスト
チームビルディング
DQ Plan /DQ Report
IQ Plan/IQ Report
OQ Plan/OQ Report
PQ Plan/PQ Report
PPQ Plan / PPQ Report
7.バリデーションのアウトプット
QC工程表と管理図
工程能力
再バリデーションの時期設定
設備メンテナンス手順
( 内容が変更になる場合もございますが大きな変更はございません。 )

2016_6
FDA GMP(QSR)2日間コース Rev.17
〜 医療機器・IVDに求められる規制要求事項の本質を学ぶ実践コース 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
12月開催 FDA 内部監査員コースに出席される場合の要件コースです
設計管理コースを受講される方もこのコースを受講されることをお勧め致します
☆ 開催概要 ☆
* FDA 品質システム規制 QSR 対訳本( 第2版 )付き
グローバルに進出しようとする企業にとって 米国は最も大きなマーケットであり、米国市場での成功がグローバルを制するといっても過言ではありません。しかし、米国に上市するには医療機器規制QSRに基づくFDAの査察は避けられません。また、医療機器を国内で販売する際にもFDA QSRのシステムフレームワーク、エビデンスベースの考え方を学び取り入れる事は、リスクの低減、新しいISO13485にも準拠し、特に受託設計、製造会社 ( OEM ) では会社のプレゼンスを高め、サプライヤ−としての評価を高めます。 クオリス・イノーバのQSRコースは、多くのFDA査察準備、対応の経験や品質管理の経験から、FDA規制のみならず医療機器開発・製造の本質を鋭く説き、多くの企業の皆様の共感を集め支持されて参りました。米国に進出する企業のみならず、医療機器・IVD を開発しようとしている企業の皆様のマインドが大きく変わる価値あるクオリス・イノーバのメインコースです。
オンサイトでも可能です。お問い合わせはこちらから。
日 時:2016年11月21日(月)、22日(火) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
OEMとして設計・製造を受託している企業
受講料: ¥99,000( 2日分、テキスト・対訳本・昼食代含む )
割 引: ¥85,000( 割引適用、テキスト・対訳本・昼食代含む )
1社2名様以上ご参加の場合
特 割: 企業で行うオンサイトセミナー開催の後、継続研修などでご利用の場合、
特別割引を適用させて頂きますのでメールでご相談下さい。
この場合、正規料金をお支払いの方が優先となりご希望に添えない場合が
ございますので予めご了承ください。
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
第1日目 セミナー内容
A.FDA規制、査察の最新情報と傾向分析
1.FDA規制、医療機器規制の本質( 医療機器開発・製造の本質を理解する )
2.FDA査察の本質、TQMを理解する
3.なぜISOではFDA査察にパスしないのか
4.ISOマインドを変える
5.根拠に基づくエビデンスの重要性
6,QSITアプローチとは
7.医療機器規制を正しく理解する為に
8.QSR規制要求事項の本質を理解する
820.20 経営者の責任
経営者がリードするTQMを理解すると見えてくる
マネジメントレビューの本質 プロセスアプローチ
品質を改善しないリスク
820.22 品質監査
効果的な監査手法と監査報告のあり方
内部監査でなぜプロセス異常を見つけられないのか
820.25 要員
教育の重要性:教育の手法と記録、力量表、リソースとは
アウェアネストレーニングの実施
820.30 設計管理( 概要のみ:設計監理1日コースで詳細解説 )
ライフサイクル製品開発手法、ウオーターフォールモデル、
フロントローディング手法、ロバスト設計
設計検証と設計バリデーションの相違
今までISO になかった設計移管、工程設計とは
設計CAPAとは
820.40 文書管理
グロバールに通用する文書管理手法と教育
820.50 購買管理
これからの査察の潮流、サプライヤー管理とは
購買チームが品質に貢献するために
820.60 識別
混同を防止する為の識別方法とは
820.65 トレーサビリティー
体内埋込型機器だけではなく、変化点を知り品質を高める
820.80 アクセプタンスアクティビティー
3つのアクセプタンスを理解する
Receiving, In-process, and Finished device acceptance
820.86 アクセプタンス状態
状態表示する意味とは
820.90 不適合品
不適合品の処理プロセス
FDAはなぜNCを見たがるのか
グローバル用語を使用し、CAPAとのリンクを示す
820.120 機器のラベリング
リコールが一番多く査察で注目されるラベリングの出納管理
ラベリングの設計とは
ラベルとラベリングの相違
820.130 機器の梱包
梱包箱を見ればどんな設計を行っていたかが判る
設計依託先はISOで十分か?
820.140 取扱い
混同・劣化・損傷・汚染を発生させないためのリスク管理
820.150 保管
倉庫などの保管エリアのリスク管理
リスクアセスメントをすると。。
820.160 流通
劣化品の流通を防ぐ設計品質とは
温度指定製品の流通保証とは
820.170 据付け
インスタレーション手順書のV&V
820.180 記録
全ての活動が根拠に基づくエビデンスで保証されるための重要な活動
Objective Evidence を示すための記録管理とは
エビデンスベースの基礎を学ぶ
820.181 機器原簿(DMR)
DMRを定義し識別する
DHF,DMR,DHRの相違
820.184 機器履歴簿(DHR)
製造記録の記載方法
コンプライアンス上、特に重要なDHR
820.186 品質システム記録
DHF,DMR,DHR以外の品質記録とは
820.200 付帯サービス
サービス記録の分析とMR,MDRの判定
820.250 統計的手法
TQMを進めていく上で最も重要な要素
品質改善に欠かせない統計的手法
第2日目 セミナー内容
820.70 生産及び工程管理
工程設計と工程V&V
工程のリスクマネジメント(PFMEA)
工程を監視、管理する工程パラメータとは
要員、環境、汚染管理
設備の妥当性とは
保守と検査の相違
副資材の定義と管理
820.72 検査・測定及び試験装置
意図した用途への適合性、測定器の精度管理とは
測定器の精度管理と救済処置
820.75 工程の妥当性確認
QSR,医療機器規制の本質、工程バリデーションとは
TQMが目指すべき試験レスとは
バリデーションの手法( VMP,DQ、IQ、OQ、PQ、PPQ )
設備バリデーションとは
CSV( コンピュータシステムバリデーション )とは
820.198 苦情ファイル
査察のキーポイント、苦情管理の目的を理解する
苦情処理とMDR( Part 803 )、CAR( Part 806 )
820.100 是正処置及び予防処置( CAPA )
FDA警告書で最も多い指摘CAPAの原則を理解する
修正、是正、予防の相違を正確に理解しISOマインドを変える
CAPA7ステップを理解する
CAPAの効果(再発防止)を出す仕組み
MRで監視すべきCAPAプロセス
( 内容が変更になる場合もございますが大きな変更はございません。)
2016_5 QSR Seminar

医療機器ソフトウェアバリデーションコース
〜 医療機器ソフトウェア開発手順の作成方法 〜
IEC 62304 Ed.1.1:2015 + FDAガイダンス
このコースの売り上げ金の一部は人道支援活動に寄付されます
☆ 開催概要 ☆
ほとんどの日本企業に於ける開発・設計管理の仕組みの中で、ソフトウェア開発については認証機関から指摘が無いのが実情です。しかし、厳しい規制当局であるFDAはソフトウェアのバグが関連する医療事故リスクを統計的に理解しており、IEC規格があってもなおガイダンスを無効にしておらず、今後査察の重要ポイントとなってきます。 一方、欧州・国内にあっては、医療機器ソフトウェアについては IEC62304 : 2006 / Ed.1.1 : 2015 規格への対応を求められています。 この IEC62304 : 2006 / Ed.1.1 : 2015 規格については、FDAのガイダンスが求める要求のサブセットとも考えられます。そこで、この差分への対応を明らかにし、FDAガイダンスを取り入れたグローバル対応の IEC62304 : 2006 / Ed.1.1 : 2015 規格ベース医療機器ソフトウェア開発手順書構築を解説致します。
日 時:2016年9月12日(月曜) 10:00 〜 16:30
予定を変更し延期となりました。 日程は追って発表致します。
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
講 師:テクマトリックス株式会社
システムエンジニアリング事業部
フェロー シニアコンサルタント 工学博士 中島 裕生 氏
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している企業
FDA査察準備を考えている企業
既にFDA査察を終え対応に困っている企業
受講料: 54,000( テキスト・昼食代含む )
割 引: 43,000( テキスト・昼食代含む )
1社2名以上受講される場合
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
このコースを受講される前に弊社設計コースを受講されることをお勧め致します。
1.FDAガイダンスを取り入れた62304ベースの手順書作成
FDAが考えるソフトウェア
ソフトウェアバリデーションに関する規格書、ガイダンス、TRについて
組込ソフトウェア医療機器とソフトウェア単体医療機器、および
品質マネジメントシステムQMSとの関係
システムライフサイクルとソフトウェア(開発/保守)ライフサイクル、および
ライフサイクルモデルの関係
V&Vとは?
リスクマネジメント
計画とレビュー
構成マネジメント
ディフェクトマネジメント
トレーサビリティ
<医療機器ソフトウェアバリデーション>
コンセプトアクティビティ
ソフトウェア要求アクティビティ
設計アクティビティ
実装アクティビティ
テストアクティビティ
保守とソフトウェア変更アクティビティ
2.FDA査察警告書から学ぶ査察の要点
FDAは何を見ているか
3.ライフサイクルマイルストーンのどの段階から検証記録を残すか
ライフサイクルマイルストーンへのソフトウェアマイルストーンの組み込み
4.FDA査察に対応する記録の残し方
構成管理と文書管理
*コース内容は一部変更される場合もございますが、大きな変更はございません。
FDA MDR・CAR コース
〜 指摘の多いMDR・リコール規制の理解とプロセス構築 〜
Reference 21CFR : Part 803, 806
このコースの売り上げ金の一部は人道支援活動に寄付されます
☆ 開催概要 ☆
最近の日本でのFDA査察で最も多い指摘はMDR(有害事象報告)です。これはMDR報告の判断に迷ったら報告するという米国の文化とは異なり、日本のメーカーは情報を積極的に発信しない、隠すという古い会社の文化が根強くあることが要因の一つですが、そもそも規制を理解せず、正しい判断を行っていないことが大きな原因です。医療機器を作っているメーカーは、患者さんの命を守るという社会的な責任があります。会社を守っているようでも、結果的に会社が倒産に追い込まれることもある非常にリスクの高いプロセスであることを理解すべきです。それには規制の正しい理解、規制を守る仕組みと組織が必要です。このセミナーでは、FDAの言うCAPAサブシステム(苦情、不適合、CAPA)の各プロセスとリンクし、日本のメーカーの Warning Letter (警告書)を引き合いに正しいMDRプロセスと関連するリコールプロセスを構築することを理解します。このコースはオンサイトでも実施しています。お問い合わせはこちらから。
日 時:2016年9月5日(月曜)10:00 〜 16:00
( 7月11日開催予定でしたが9月5日に延期となりました )
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:既に医療機器・IVDを海外に輸出している企業
既にFDA査察を終え対応に困っている企業
受講料: ¥50,000
割 引: ¥40,000 1社2名以上受講される場合
定 員: 16名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
1.規制を理解する
事例と統計からみる規制違反
理解すべき規制・規格・ガイダンス
eMDR規制
規制が求めるあるべき組織と力量
用語の定義を理解する
2.規制当局が考えているプロセスとは
FDAが理解しているサブシステムとは
苦情処理、CAPA、不適合処理、リコールプロセスとのリンク
社内苦情と顧客からの苦情
規制で求められる契約書
3.MDRプロセス
苦情処理プロセス
不適合処理プロセス
CAPAプロセス
MDR報告プロセス eMDR
ディシジョンツリー
4.CAR( リコール )プロセス
改修及び回収
用語の定義を理解する
報告義務のないCARとは
CARプロセス
5.警告書から理解すべき注意点とプロセスへの展開
日本の Warning Letter ( 警告書 )の事例から理解する
警告書から理解すべき注意点とプロセスへの展開
*コース内容が一部変更される場合がございますが大きな変更はございません。
FDA QSR 内部監査員コース
〜 QSR 対応内部監査員養成のための社内資格認定2日間コース 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
締め切りました。次回のご出席をお待ちしております!
☆ 開催概要 ☆
不適合を出さないようプロセスに内在する不適合を未然に抽出し、マネジメントレビューでトップマネジメントの指示のもとプロセス改善を常に行う。これがTQMの有るべき姿であり規制当局が求めている姿です。一方、現実はプロセスを監視し傾向管理を行っておらず、よく訓練されていないにわか監査員がISOで要求されているからといって片手間に監査を行っているだけ。これは医療機器の場合致命的で、リコールで何十億の損失を出すだけではなく人の命に関わると会社の存続まで危ぶまれます。このコースでは、ISO内部監査員の資格を持つ方々がQSRの内部監査が行えるよう社内資格を付与するための社内資格認定コースとして位置づけられます。一般に行われているISOの内部監査手法とは異なる、付加価値を生み、予防対策としての監査手法とノウハウを学びます。( このコースを受講した後、オンサイトでクオリス・イノーバによる内部監査のOJTを行うとより効果的です )
日 時:2016年7月19日(火曜)、20日(水曜) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:すでに弊社主催下記コースをオープン、オンサイト問わず受講された方限定です。
* FDA GMP QSR 2日間コース
* FDA 設計管理2日間コース
このコースはQSRの基礎知識が無いと受講できません。弊社のコースを受講されていない
場合、異なる解釈により内容をよく理解出来ません。 皆様には何卒ご理解をお願い申し上
げます。
受講料: ¥99,000( テキスト、昼食代含む、内税 )
このコースには割引がございません。
定 員: 18名限定 満席になり次第締切ます
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
過去に受講された弊社QSR、設計管理各コースの受講日を記載して下さい。
オンサイトで受講された場合でも構いません。
折り返し、会場、お振込先情報をメールでご連絡致します。
![]()
お問い合わせ: お問い合わせのページからお問い合わせ下さい
お電話の方は下記セミナー事務局までお問い合わせ下さい
042−856−2208
☆ セミナー内容 ☆
1.内部監査の目的を理解する
規制・規格が求めるていること
患者の命を優先する監査とは
企業の付加価値を高める為には
2.内部監査員に求められる力量
内部監査員とリーダーに求められる力量
内部監査員トレーニング
マネジメントチームのカウンセラー
3.プロセスアプローチ
CAPAの理解とCAPAアプローチ
プロセスのR&Rを明確にする
プロセスを常に測定し改善する
内部監査はプロセスに注目
4.監査判定基準
チェックリストと数値化
プロセス毎に数値化
FDA査察と内部監査判断基準の相違を知る
5.監査を計画する
マネジメントレビューと品質会議
プロセス管理パラメートと傾向分析結果
是正監査と予防監査の相違
監査は短期集中型で実施
監査員レビュー会のノウハウ
監査報告書記載のポイント
6.監査の目のつけどころ( QSIT )
QSITに基づく監査手法と勘所
1.経営者による管理
2.設計管理
3.CAPA
MDR報告
CAR
MDトラッキング
4.P&PC
5.滅菌工程
*内容は変更される場合がございます*
前回講習風景 2015

FDA設計管理2日間コース Rev.16
〜 規制だけではなく会社に利益をもたらすものづくりの本質 〜
Reference 21CFR: 820 Subpart C, G
このコースの売り上げ金の一部は人道支援活動に寄付されます
7月開催 FDA 内部監査員コースの出席条件となるコースです
今回から設計移管( 工程設計・プロセスバリデーション )を含む2日間コースとなりました
締め切りました 次回のご参加を待ちしております!
☆ 開催概要 ☆
ほとんどの日本企業に於ける開発・設計管理の仕組みは、医療機器設計の世界標準と言っても過言ではない米国FDA規制に準拠していません。これはグローバルに進出しようとする企業だけではなく、患者の命を預かる全ての医療機器メーカーにとって大きなリスクを伴います。FDAは自らの査察で医療機器品質不良の根本原因は設計管理にあると統計的に見抜いており、それはクオリス・イノーバの多くのGAP監査経験でも明かです。そこで本コースでは、グローバルな開発・設計管理を「FDA設計管理ガイダンス」と、欧米で取り入れられているフレームワークを元に体系的に解説し、医療機器開発の本質である品質不良を発生させない仕組み(ロバスト設計)をタグチメソッドと設計CAPAを用いて解説します。ただ ” 規制だから " ではなく、設計品質を高めると企業にとっても大きな潜在的利益を得られ、エンジニアの方から多くの共感と支持を集めてきたクオリス・イノーバの最も人気のある価値あるコースです。 このコースはオンサイトでも実施しています。お問い合わせはこちらから。
日 時:2016年6月13日(月曜)、14日(火曜) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している企業
FDA査察準備を考えている企業
既にFDA査察を終え対応に困っている企業
受講料: ¥99,000( テキスト・昼食代含む )
割 引: ¥85,000( テキスト・昼食代含む )
1社2名以上受講される場合
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
このコースを受講される前にQSR2日コースを受講されることをお勧め致します。
弊社主催 内部監査員コースは、QSR2日間コースと設計監理2日間コースの両方の出席が要件です。
第1日目 セミナー内容
1.設計品質の本質的価値
繰り返される設計品質に起因する製品不良
ISOとQSRの本質的な違い
理解すべき規制・規格・ガイダンス
FDAが望んでいるTQMの本質、品質工学に基づくロバスト設計
医療機器の設計手法( Water Fall Model )
一貫性を問うFDA査察
〜 TQM、FDA設計管理ガイダンスに基づくQSRサブパートCの実践 〜
2.820.30 ( a ) 総則
TQMに欠かせないマネジメントチームの重要な役割
設計環境、開発リソースの見極め
要員教育の重要性( 820.25 )
力量表と教育計画
3.820.30 ( b ) 設計及び開発計画
医療機器開発ライフサイクルマネジメント
リスクマネジメントライフサイクル
タグチメソッドの開発方式によるフロントローディング手法
PM,PLの役割
カスタマーニーズを要求仕様に変換する品質機能展開
設計品質確立の重要な要素、コンカレントエンジニアリング
設計計画書とは
4.820.30 ( c ) 設計インプット
ロバスト設計( タグチメソッド )による未然防止設計
要求仕様書から設計仕様書への変換
曖昧なニーズのスペックへの変換
重要な設計要素トレーサビリティーマトリックスの使用
ラベリング、梱包の設計インプット
5.820.30 ( d ) 設計アウトプット
入力のバラツキを押さえたアウトプットの確認
設計アウトプットを定義し文書類を明確にする
E-BOM / M-BOM とは
製造仕様書とは
図面発行
DMRとのトレーサビリティー
6.820.30 ( j ) 設計履歴ファイル( DHF )
DHF / DMR / DHR の定義を理解する
識別、ファイルの基本
文書体系と設計基準
7.820.30 ( e ) 設計審査
問題の早期発見、解決が目的で、セレモニーで決して終わらない
設計審査で設計のロバストネスを確認
設計審査とテクニカルレビュー
キーとなる審査者
8.820.30 ( f ) 設計検証
設計検証と設計バリデーションの相違
インプットからアウトプットまでトレーサブルであるとは
エビデンスに基づく設計
設計検証レポートは、設計者の活動記録
9.820.30 ( g ) 設計バリデーション
設計バリデーションの手法
社内、臨床評価の相違と記録
ソフトウェアバリデーションとVモデル
3種類のバリデーションの相違
10.820.30 ( i ) 設計変更
重要な査察ポイント
設計変更プロセスとリスクアセスメント
コミュニケーションエラー
製品不良を再発させない重要な要素、設計CAPA
文書、図面の変更管理
( 内容が変更になる場合もございますが大きな変更はございません。 )
第2日目 セミナー内容
1.なぜ工程バリデーションを実施するのか?
プロセスバリデーションの目的とその本質
プロセスバリデーションの効果
工程管理の目的と本質
医療機器規制・規格を理解する
820.30(h)設計移管
2.バリデーションすべき工程とは
バリデーションすべき工程の識別
検証工程とバリデーション工程の違い
設備バリデーションと工程バリデーション
バリデーション工程の例
3.バリデーションの種類
バリデーションの種類
設備バリデーションと劣化パラメータ
コンピュータシステムバリデーション(CSV)
4.工程設計とバリデーションステップ
VMP: バリデーションマスタープラン
DQ: 設計時的確性評価
IQ: 据付時的確性評価
OQ: 稼働時的確性評価
PQ: 製造性能的確性評価
PPQ
バリデーションの文書化
プロセスバリデーションの明確化
自動化工程の明確化
CSVカテゴリーの明確化
ソフトウェアバリデーションの基本
カテゴリー別ソフトウェア
5.設計移管とプロセスバリデーション
設計移管とプロセスバリデーションの関係
ミニスケールとフルスケール
工程設計表とトレーサビリティー
6.バリデーションの実践
マスターバリデーションプラン
バリデーション工程の識別
バリデーション計画
設備リストとバリデーション工程リスト
チームビルディング
DQ Plan /DQ Report
IQ Plan/IQ Report
OQ Plan/OQ Report
PQ Plan/PQ Report
PPQ Plan / PPQ Report
7.バリデーションのアウトプット
QC工程表と管理図
工程能力
再バリデーションの時期設定
設備メンテナンス手順
2015_6

FDA GMP(QSR)2日間コース Rev.16
〜 医療機器・IVDに求められる規制要求事項の本質を学ぶ実践コース 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
7月開催 FDA 内部監査員コースの出席条件となるコースです
設計管理コースを受講される方もこのコースを受講されることをお勧め致します
定員オーバーにつき、募集を締め切りました
次回のご参加をお待ちしております
☆ 開催概要 ☆
* FDA 品質システム規制 QSR 対訳本( 第2版 )付き
グローバルに進出しようとする企業にとって 米国は最も大きなマーケットであり、米国市場での成功がグローバルを制するといっても過言ではありません。しかし、米国に上市するには医療機器規制QSRに基づくFDAの査察は避けられません。また、医療機器を国内で販売する際にもFDA QSRのシステムフレームワーク、エビデンスベースの考え方を学び取り入れる事は、リスクの低減、新しいISO13485にも準拠し、特に受託設計、製造会社 ( OEM ) では会社のプレゼンスを高め、サプライヤ−としての評価を高めます。 クオリス・イノーバのQSRコースは、多くのFDA査察準備、対応の経験や品質管理の経験から、FDA規制のみならず医療機器開発・製造の本質を鋭く説き、多くの企業の皆様の共感を集め支持されて参りました。米国に進出する企業のみならず、医療機器・IVD を開発しようとしている企業の皆様のマインドが大きく変わる価値あるクオリス・イノーバのメインコースです。
オンサイトでも可能です。お問い合わせはこちらから。
日 時:2016年5月16日(月)、17日(火) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
OEMとして設計・製造を受託している企業
受講料: ¥99,000( 2日分、テキスト・対訳本・昼食代含む )
割 引: ¥85,000( 割引適用、テキスト・対訳本・昼食代含む )
1社2名様以上ご参加の場合
特 割: 企業で行うオンサイトセミナー開催の後、継続研修などでご利用の場合、
特別割引を適用させて頂きますのでメールでご相談下さい。
この場合、正規料金をお支払いの方が優先となりご希望に添えない場合が
ございますので予めご了承ください。
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
第1日目 セミナー内容
A.FDA規制、査察の最新情報と傾向分析
1.FDA規制、医療機器規制の本質( 医療機器開発・製造の本質を理解する )
2.FDA査察の本質、TQMを理解する
3.なぜISOではFDA査察にパスしないのか
4.ISOマインドを変える
5.根拠に基づくエビデンスの重要性
6,QSITアプローチとは
7.医療機器規制を正しく理解する為に
8.QSR規制要求事項の本質を理解する
820.20 経営者の責任
品質目標展開と品質計画
経営者がリードするTQMを理解すると見えてくる
マネジメントレビューの本質
FDA が一番確認したいこととは。。
820.22 品質監査
効果的な監査手法と監査報告のあり方
内部監査員のFDAエキスパートの相違
820.25 要員
教育の重要性:教育の手法と記録、力量表、リソースとは
アウェアネストレーニングの実施
820.30 設計管理( 概要のみ:設計監理1日コースで詳細解説 )
ライフサイクル製品開発手法、ウオーターフォールモデル、
フロントローディング手法、ロバスト設計
設計検証と設計バリデーションの相違
ISO にない設計移管、工程設計とは
設計CAPAとは
820.40 文書管理
グロバールに通用する文書管理手法と教育
820.50 購買管理
これからの査察の潮流、サプライヤー管理とは
購買チームが品質に貢献するために
820.60 識別
混同を防止する為の識別方法とは
820.65 トレーサビリティー
体内埋込型機器だけではなく、変化点を知り品質を高める
820.80 受領、工程内及び完成機器のアクセプタンス
3つのアクセプタンスを理解する
Receiving, In-process, and Finished device acceptance の相違
820.86 アクセプタンス状態
状態表示する意味とは
820.90 不適合品
不適合品の処理プロセス
FDAはなぜNCを見たがるのか
グローバル用語を使用し、CAPAとのリンクを示す
820.120 機器のラベリング
リコールが一番多く査察で注目されるラベリングの出納管理、
ラベリングの設計とは
ラベルとラベリングの相違
820.130 機器の梱包
梱包箱を見ればどんな設計を行っていたかが判る
設計依託先はISOで十分か?
820.140 取扱い
混同・劣化・損傷・汚染を発生させないためのリスク管理
820.150 保管
倉庫などの保管エリアのリスク管理
リスクアセスメントをすると。。
820.160 流通
劣化品の流通を防ぐ設計品質とは
温度指定製品の流通保証とは
820.170 据付け
インスタレーション手順書のV&V
820.180 記録
全ての活動が根拠に基づくエビデンスで保証されるための重要な活動
Objective Evidence を示すための記録管理とは
エビデンスベースの基礎を学ぶ
820.181 機器原簿(DMR)
DMRを定義し識別する
DHF,DMR,DHRの相違
820.184 機器履歴簿(DHR)
製造記録の記載方法
コンプライアンス上、特に重要なDHR
820.186 品質システム記録
DHF,DMR,DHR以外の品質記録とは
820.200 付帯サービス
サービス記録の分析とMR,MDRの判定
820.250 統計的手法
TQMを進めていく上で最も重要な要素
品質改善に欠かせない統計的手法
第2日目 セミナー内容
820.70 生産及び工程管理
工程設計と工程V&V
工程のリスクマネジメント(PFMEA)
工程を監視、管理する工程パラメータとは
要員、環境、汚染管理
設備の妥当性とは
保守と検査の相違
副資材の定義と管理
820.72 検査・測定及び試験装置
意図した用途への適合性、測定器の精度管理とは
測定器の精度管理と救済処置
820.75 工程の妥当性確認
QSR,医療機器規制の本質、工程バリデーションとは
TQMが目指すべき試験レスとは
バリデーションの手法( VMP,DQ、IQ、OQ、PQ、PPQ )
設備バリデーションとは
CSV( コンピュータシステムバリデーション )とは
820.198 苦情ファイル
査察のキーポイント、苦情管理の目的を理解する
苦情処理とMDR( Part 803 )、CAR( Part 806 )
820.100 是正処置及び予防処置( CAPA )
FDA警告書で最も多い指摘CAPAの原則を理解する
修正、是正、予防の相違を正確に理解しISOマインドを変える
CAPA7ステップを理解する
CAPAの効果(再発防止)を出す仕組み
MRで監視すべきCAPAプロセス
( 内容が変更になる場合もございますが大きな変更はございません。)
2015_11 QSR Seminar

プロセスバリデーションコース Rev.11
〜 プロセスバリデーションの本質の理解と工程設計 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
定員オーバーにつき募集を締め切りました
次回の参加をお待ちしています
☆ 開催概要 ☆
品質システムが目指すものは、意図した用途に適合する製品を終始一貫して生産すること。これらの原則と目的を達成するための重要な要素がプロセスバリデーションです。そこで、規制当局はプロセスバリデーションの手順と結果を確認しようとします。FDA査察の結果を統計的にみてもワースト10に入る指摘項目となっています。クオリス・イノーバがこれまで実施してきたGAP監査でもプロセスバリデーションのエビデンスが提出できず手順さえもない企業がほとんどです。エビデンスを提出できたとしてもプロセスバリデーションの本質を理解していないので、監査に耐えられる満足のいくものではありません。このコースではISOにはない設計移管から工程設計、マスタープランからCSVを含むプロセスバリデーションを解説、製品品質のバラツキを無くして医療機器の製品安全性に確保し、品質コストを下げるプロセスバリデーションの本質を理解します。
* Rev.11 では工程設計と共にプロセスバリデーションを理解するよう改訂されています
日 時:2016年3月14日(月曜) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している企業
FDA査察準備を考えている企業
受講料: ¥50,000
割 引: ¥40,000
1社2名以上ご参加の場合
定 員: 20名 (3月8日締切)
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
特 割: プロジェクトで実施するオンサイトセミナーの後、継続研修などでご利用戴く場合、
特別割引をさせて戴きますのでお問い合わせ下さい。 この場合、正規料金をお支払
いのお客様が優先され御出席できない場合がございますので予めご了解下さい。
再受講: 再度受講される場合、ご負担を軽減するために特別割引を設定しています。
うまくいかない点を解決しプロセスの理解を深める機会にご利用下さい。
この場合、空席がある場合にご参加戴けます。お申込の際、再受講とご記入
され、併せて受講された日もご記入下さい。
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
![]()
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
1.なぜ工程バリデーションを実施するのか?
プロセスバリデーションの目的とその本質
プロセスバリデーションの効果
工程管理の目的と本質
医療機器規制・規格を理解する
2.バリデーションすべき工程とは
バリデーションすべき工程の識別
検証工程とバリデーション工程の違い
設備バリデーションと工程バリデーション
バリデーション工程の例
3.バリデーションの種類
バリデーションの種類
設備バリデーションと劣化パラメータ
コンピュータシステムバリデーション(CSV)
4.工程設計とバリデーションステップ
VMP: バリデーションマスタープラン
DQ: 設計時的確性評価
IQ: 据付時的確性評価
OQ: 稼働時的確性評価
PQ: 製造性能的確性評価
PPQ
バリデーションの文書化
プロセスバリデーションの明確化
自動化工程の明確化
CSVカテゴリーの明確化
ソフトウェアバリデーションの基本
カテゴリー別ソフトウェア
5.設計移管とプロセスバリデーション
設計移管とプロセスバリデーションの関係
ミニスケールとフルスケール
工程設計表とトレーサビリティー
6.バリデーションの実践
マスターバリデーションプラン
バリデーション工程の識別
バリデーション計画
設備リストとバリデーション工程リスト
チームビルディング
DQ Plan /DQ Report
IQ Plan/IQ Report
OQ Plan/OQ Report
PQ Plan/PQ Report
PPQ Plan / PPQ Report
7.バリデーションのアウトプット
QC工程表と管理図
工程能力
再バリデーションの時期設定
設備メンテナンス手順
*内容は変更される場合がございます*

CAPA コース Rev.15
〜 査察で最も指摘の多いCAPAの正しい理解と問題を再発させない仕組み 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
定員オーバーにつき募集を締め切りました
次回のご参加をお待ちしております
☆ 開催概要 ☆
FDA査察の結果を統計的にまとめてみるとCAPAで最も多くの指摘をしていることが判ります。これは日本企業の指摘でも顕著です。FDAは自らの査察を通して企業で最も理解されていないプロセスを統計的によく理解しており、不適合の再発原因がCAPAプロセスにあることを知っています。このことはクオリス・イノーバが実施してきたGAP監査でも明かで、日本のほとんどの企業でCAPAを正しく理解しておらずマネジメントできていません。これは品質管理が行われていないことを如実に示しており、医療の現場では非常にリスクです。また、FDAの査察を受けた後のアクションレターもCAPAのステップで回答すべきです。 このコースに参加指されたほとんどの方のCAPAのマインドが変わる価値あるコースです。 医療機器だけではなく、医薬品や多くの業種で理解すべき内容です。
* 今回の Rev.15 では ケーススタディーを多く取り入ています
日 時:2016年3月7日(月曜) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVDに限らず医薬品などCAPAを用いた品質改善を期待される企業
受講料: ¥50,000( テキスト・昼食代含む )
割 引: ¥40,000( テキスト・昼食代含む )
1社2名以上ご参加の場合
特 割: プロジェクトで実施するオンサイトセミナーの後、継続研修などでご利用戴く場合、
特別割引をさせて戴きますのでお問い合わせ下さい。 この場合、正規料金をお支払
いのお客様が優先され御出席できない場合がございますので予めご了解下さい。
再受講: 席に空きがある場合、特別料金で受講出来ます。この機会にご参加なさり、
うまくいかない点を解決しプロセスの理解を深めて下さい。
お申込の際、再受講とご記入され、併せて受講された日もご記入下さい。
定 員: 20名 3月1日締切
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
![]()
☆ セミナー内容 ☆
1.なぜFDAはCAPAに注目するのか
2.なぜ品質問題は再発するのか
3.規制・規格を理解する
FDA 21CFR Part 820.100 CAPA vs. ISO
4.TQMの仕組みが不可欠
マネジメントチームの理解不足が品質不良を招く
5.用語を正しく理解する
修正、是正、予防の相違
不適合、逸脱、特別採用と逸脱許可の相違
修正、手直し、修理の相違
6.直接原因と根本原因を区別する
この違いの理解が最も重要
プロセスアプローチとは
7.修正、是正のプロセス
苦情処理、不適合処理、CAPA手順との関係
8.予防プロセス
品質管理の原則は予防活動
9.CAPAエスカレーションプロセス
CAPAプロセスをうまくまわす為に
10.CAPA7ステップ
Six Sigma との相違
Step 1 問題を検出し明確にする
Step 2 問題の原因を詳しく調査する
Step 3 根本原因を特定する
Step 4 是正又は予防するための処置
Step 5 処置の結果を文書化
Step 6 処置の検証・妥当性確認
Step 7 有効性の確認
11.CR/CARの実施
12.製品開発にCAPAを活かす
13.CAPAのマネジメント
14.品質問題の事例に学ぶ
Warning Letter Case Study
実例に学ぶ
* 内容が一部変更となる場合がございます

FDA QSR 内部監査員コース
〜 QSR 対応内部監査員養成のための社内資格認定2日間コース 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
募集を締め切りました
次回のご参加をお待ちしています
☆ 開催概要 ☆
不適合を出さないようプロセスに内在する不適合を未然に抽出し、マネジメントレビューでトップマネジメントの指示のもとプロセス改善を常に行う。これがTQMの有るべき姿であり規制当局が求めている姿です。一方、現実はプロセスを監視し傾向管理を行っておらず、よく訓練されていないにわか監査員がISOで要求されているからといって片手間に監査を行っているだけ。これは医療機器の場合致命的で、リコールで何十億の損失を出すだけではなく人の命に関わると会社の存続まで危ぶまれます。このコースでは、ISO内部監査員の資格を持つ方々がQSRの内部監査が行えるよう社内資格を付与するための社内資格認定コースとして位置づけられます。一般に行われているISOの内部監査手法とは異なる、付加価値を生み、予防対策としての監査手法とノウハウを学びます。( このコースを受講した後、オンサイトでクオリス・イノーバによる内部監査のOJTを行うとより効果的です )
* 今回より2日コースとなりました *
日 時:2015年12月14日(月曜)、15日(火曜) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:すでに弊社主催下記コースをオープン、オンサイト問わず受講された方限定です。
* FDA GMP QSR 2日間コース
* FDA 設計管理コース
このコースはQSRの基礎知識が無いと受講できません。弊社のコースを受講されていない
場合、異なる解釈により内容をよく理解出来ません。 皆様には何卒ご理解をお願い申し上
げます。
受講料: ¥98,000( テキスト、対訳本、QSIT邦訳、昼食代含む、内税 )
このコースは割引がございません。
定 員: 20名限定 満席になり次第締切ます
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
過去に受講された弊社QSR2日間コースと設計管理コースの受講日を記載して下さい。
オンサイトで受講された場合でも構いません。
折り返し、会場、お振込先情報をメールでご連絡致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
1.内部監査の目的を理解する
規制・規格が求めるていること
患者の命を優先する監査とは
企業の付加価値を高める為には
2.内部監査員に求められる力量
内部監査員とリーダーに求められる力量
内部監査員トレーニング
マネジメントチームのカウンセラー
3.プロセスアプローチ
CAPAの理解とCAPAアプローチ
プロセスのR&Rを明確にする
プロセスを常に測定し改善する
内部監査はプロセスに注目
4.監査判定基準
チェックリストと数値化
プロセス毎に数値化
FDA査察と内部監査判断基準の相違を知る
5.監査を計画する
マネジメントレビューと品質会議
プロセス管理パラメートと傾向分析結果
是正監査と予防監査の相違
監査は短期集中型で実施
監査員レビュー会のノウハウ
監査報告書記載のポイント
6.監査の目のつけどころ( QSIT )
QSITに基づく監査手法と勘所
1.経営者による管理
2.設計管理
3.CAPA
MDR報告
CAR
MDトラッキング
4.P&PC
5.滅菌工程
*内容は変更される場合がございます*
前回講習風景

FDA設計管理ガイダンスコース Rev.15
〜 規制だけではなく会社に利益をもたらすものづくりの本質 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
募集を締め切りました
次回のご参加をお待ちしています
☆ 開催概要 ☆
ほとんどの日本企業に於ける開発・設計管理の仕組みは、米国FDAが要求する規制に準拠していません。これはグローバルに進出しようとする企業だけではなく、患者の命を預かる全ての医療機器メーカーにとって大きなリスクを伴います。FDAは自らの査察で医療機器品質不良の根本原因は設計管理にあると統計的に見抜いており、それはクオリス・イノーバの多くのGAP監査経験でも明かです。そこで本コースでは、グローバルな開発・設計管理を「FDA設計管理ガイダンス」と、欧米で取り入れられているフレームワークを元に体系的に解説し、医療機器開発の本質である品質不良を発生させない仕組み(ロバスト設計)をタグチメソッドと設計CAPAを用いて解説します。ただ規制だからではなく、設計品質を高めると、企業にとっても大きな潜在的利益を得られます。エンジニアの方から多くの共感と支持を集めてきたクオリス・イノーバの最も人気のある価値あるコースです。 オンサイトでも実施しています。お問い合わせはこちらから。
日 時:2015年11月24日(火曜) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している企業
FDA査察準備を考えている企業
既にFDA査察を終え対応に困っている企業
受講料: ¥50,000( テキスト・規制邦訳文・昼食代含む )
割 引: ¥40,000( テキスト・規制邦訳文・昼食代含む )
1社2名以上ご参加の場合、或いは2015年QSRコース(11月9日、10日)を
受講された方
特 割: オンサイトセミナーの後、継続研修などでご利用の場合、特別割引を適用させて頂
きますのでご相談下さい。 この場合、正規料金をお支払い頂く方が優先され、
人気コースの為ご出席戴けない場合がございますので予めご了解下さい。
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
このコースを受講される前にQSR2日コースを受講されることをお勧め致します。
弊社主催 内部監査員コースは、QSR2日間コースと設計監理コースの両方の出席が要件です。
1.設計品質の本質的価値
繰り返される設計品質に起因する製品不良
ISOとQSRの本質的な違い
理解すべき規制・規格・ガイダンス
FDAが望んでいるTQMの本質、品質工学に基づくロバスト設計
医療機器の設計手法( Water Fall Model )
一貫性を問うFDA査察
〜 TQM、FDA設計管理ガイダンスに基づくQSRサブパートCの実践 〜
2.820.30 ( a ) 総則
TQMに欠かせないマネジメントチームの重要な役割
設計環境、開発リソースの見極め
要員教育の重要性( 820.25 )
力量表と教育計画
3.820.30 ( b ) 設計及び開発計画
医療機器開発ライフサイクルマネジメント
リスクマネジメントライフサイクル
タグチメソッドの開発方式によるフロントローディング手法
PM,PLの役割
カスタマーニーズを要求仕様に変換する品質機能展開
設計品質確立の重要な要素、コンカレントエンジニアリング
設計計画書とは
4.820.30 ( c ) 設計インプット
ロバスト設計( タグチメソッド )による未然防止設計
要求仕様書から設計仕様書への変換
曖昧なニーズのスペックへの変換
重要な設計要素トレーサビリティーマトリックスの使用
ラベリング、梱包の設計インプット
5.820.30 ( d ) 設計アウトプット
入力のバラツキを押さえたアウトプットの確認
設計アウトプットを定義し文書類を明確にする
E-BOM / M-BOM とは
製造仕様書とは
図面発行
DMRとのトレーサビリティー
6.820.30 ( j ) 設計履歴ファイル( DHF )
DHF / DMR / DHR の定義を理解する
識別、ファイルの基本
文書体系と設計基準
7.820.30 ( e ) 設計審査
問題の早期発見、解決が目的で、セレモニーで決して終わらない
設計審査で設計のロバストネスを確認
設計審査とテクニカルレビュー
キーとなる審査者
8.820.30 ( f ) 設計検証
設計検証と設計バリデーションの相違
インプットからアウトプットまでトレーサブルであるとは
エビデンスに基づく設計
設計検証レポートは、設計者の活動記録
9.820.30 ( g ) 設計バリデーション
設計バリデーションの手法
社内、臨床評価の相違と記録
ソフトウェアバリデーションとVモデル
3種類のバリデーションの相違
10.820.30 ( h ) 設計移管
工程設計の基本
工程V&VとQC工程表
機能展開とPFMEA
製造仕様書への展開
DHF,DMR,DHR
11.820.30 ( i ) 設計変更
重要な査察ポイント
設計変更プロセスとリスクアセスメント
コミュニケーションエラー
製品不良を再発させない重要な要素、設計CAPA
文書、図面の変更管理
( 内容が変更になる場合もございますが大きな変更はございません。 )
2015_6

FDA GMP( QSR )2日間コース Rev.15
〜 医療機器・IVDに求められる規制要求事項の本質を学ぶ実践コース 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
定員オーバーの為、締め切りました。
次回は来年4月以降となります。
オンサイトでも可能ですのでお問い合わせ下さい。
☆ 開催概要 ☆
* QSR 対訳本付き
グローバルに進出しようとする企業にとって 米国は最も大きなマーケットであり、米国市場での成功がグローバルを制するといっても過言ではありません。しかしながら、米国に上市するには医療機器規制QSRに基づくFDAの査察は避けられません。クオリス・イノーバのQSRコースは、多くのFDA査察準備、対応の経験や品質管理の経験から、FDA規制のみならず医療機器開発・製造の本質を鋭く説き、多くの企業の皆様の共感を集め支持されて参りました。米国に進出する企業のみならず、医療機器・IVD を開発しようとしている企業の皆様のマインドが大きく変わる価値あるクオリス・イノーバのメインコースです。 オンサイトでも可能です。お問い合わせはこちらから。
日 時:2015年11月9日(月)、10日(火) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
受講料: ¥98,000( 2日分、テキスト・対訳本・昼食代含む )
割 引: ¥83,000( 2日割引適用、テキスト・対訳本・昼食代含む )
1社2名様以上ご参加の場合
特 割: 企業で行うオンサイトセミナー開催の後、継続研修などでご利用の場合、
特別割引を適用させて頂きますのでメールでご相談下さい。
この場合、正規料金をお支払いの方が優先となりご希望に添えない場合が
ございますので予めご了承ください。
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
2名以上でご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
第1日目 セミナー内容
A.FDA規制、査察の最新情報と傾向分析
1.FDA規制、医療機器規制の本質( 医療機器開発・製造の本質を理解する )
2.FDA査察の本質、TQMを理解する
3.なぜISOではFDA査察にパスしないのか
4.ISOマインドを変える
5.根拠に基づくエビデンスの重要性
6,QSITアプローチとは
7.医療機器規制を正しく理解する為に
8.QSR規制要求事項の本質を理解する
820.20 経営者の責任
品質目標展開と品質計画
経営者がリードするTQMを理解すると見えてくる
マネジメントレビューの本質
FDA が一番確認したいこととは。。
820.22 品質監査
効果的な監査手法と監査報告のあり方
内部監査員のFDAエキスパートの相違
820.25 要員
教育の重要性:教育の手法と記録、力量表、リソースとは
アウェアネストレーニングの実施
820.30 設計管理( 概要のみ:設計監理1日コースで詳細解説 )
ライフサイクル製品開発手法、ウオーターフォールモデル、
フロントローディング手法、ロバスト設計
設計検証と設計バリデーションの相違
ISO にない設計移管、工程設計とは
設計CAPAとは
820.40 文書管理
グロバールに通用する文書管理手法と教育
820.50 購買管理
これからの査察の潮流、サプライヤー管理とは
購買チームが品質に貢献するために
820.60 識別
混同を防止する為の識別方法とは
820.65 トレーサビリティー
体内埋込型機器だけではなく、変化点を知り品質を高める
820.80 受領、工程内及び完成機器のアクセプタンス
アクセプタンスを理解する
Receiving, In-process, and Finished device acceptance の相違
820.86 アクセプタンス状態
状態表示する意味
820.90 不適合品
不適合品の処理プロセス
FDAはなぜNCを見たがるのか
グローバル用語を使用する
820.120 機器のラベリング
リコールが一番多く、査察で注目されるラベリングの管理とは
ラベルとラベリングの相違
820.130 機器の梱包
梱包箱を見ればどんな設計を行っていたかが判る
820.140 取扱い
混同・劣化・損傷・汚染を発生させないためのリスク管理
820.150 保管
倉庫などの保管エリアのリスク管理
リスクアセスメントをすると。。
820.160 流通
劣化品の流通を防ぐ設計品質とは
820.170 据付け
インスタレーション手順書のV&V
820.180 記録
全ての活動が根拠に基づくエビデンスで保証されるための重要な活動
Objective Evidence を示すための記録管理とは
820.181 機器原簿(DMR)
DMRを定義し識別する
DHF,DMR,DHRの相違
820.184 機器履歴簿(DHR)
製造記録の管理方法、特に重要な。。
820.186 品質システム記録
DHF,DMR,DHR以外の品質記録とは
820.200 付帯サービス
サービス記録の分析とMR,MDRの判定
820.250 統計的手法
TQMを進めていく上で最も重要な要素
品質改善に欠かせない統計的手法
第2日目 セミナー内容
820.70 生産及び工程管理
工程を監視、管理する工程パラメータとは
工程設計と工程V&V
工程のリスクマネジメント(PFMEA)
要員、環境、汚染管理
設備の妥当性とは
保守と検査の相違
副資材の定義と管理
820.72 検査・測定及び試験装置
意図した用途への適合性、測定器の精度管理とは
測定器の精度管理と救済処置
820.75 工程の妥当性確認
QSR,医療機器規制の本質、工程バリデーションとは
TQMが目指すべき試験レスとは
バリデーションの手法( VMP,DQ、IQ、OQ、PQ、PPQ )
設備バリデーションとは
CSV( コンピュータシステムバリデーション )とは
820.198 苦情ファイル
査察のキーポイント、苦情管理の目的を理解する
苦情処理とMDR( Part 803 )、CAR( Part 806 )
820.100 是正処置及び予防処置( CAPA )
FDA警告書で最も多い指摘CAPAの原則を理解する
修正、是正、予防の相違を正確に理解しISOマインドを変える
CAPA7ステップを理解する
CAPAの効果(再発防止)を出す仕組み
MRで監視すべきCAPAプロセス
( 内容が変更になる場合もございますが大きな変更はございません。)
 2015_6 QSR Seminar
2015_6 QSR Seminar
医療機器新規参入の手引き
〜 製品開発の勘所を押さえる 〜
このコースは【 日経ものづくり 】主催で開催されます。
http://techon.nikkeibp.co.jp/article/SEMINAR/20150205/402675/?rt=nocnt
終了しました。
次回は来年4月です!

日経BP社インタビュー記事です
http://techon.nikkeibp.co.jp/atcl/column/15/417263/091800016/?ST=skillup
☆ 開催概要 ☆
日 時: 2015年10月2日(金曜) 13:30 〜 17:30
会 場: Learning Square 新橋 6F( 東京・新橋 )
主 催: 日経ものづくり
お申込: 主催者HPより直接お申し込み下さい
http://techon.nikkeibp.co.jp/article/SEMINAR/20150205/402675/?rt=nocnt
セミナー内容:
1.製品企画
医療機器のマーケティングとは
設計バリデーションの基本、カスタマーニーズと意図した用途を理解する
製品のライフサイクルマイルストーンを確立する
日本企業が知らない海外医療器機器メーカーの買収リスク
2.製品開発プロセス
医療機器はグローバル対応の設計フレームワークが必須
医療機器開発の基本、ウオーターフォールモデル
理解されていない重要な要素、設計検証と設計バリデーションの相違
要求仕様から勢品詞要所までの一貫性を示すトレーサビリティーマトリックスとは
全ての活動を根拠に基づくエビデンスで保証することの重要性
3.品質、製品不具合
トヨタ自動車のリコール問題から学ぶトップマネジメントの関わり方
テルモの出荷停止・罰金問題から学ぶマネジメントチームの関わり方
米国食品医薬品局(FDA)の警告書から判る会社の欠陥
患者や企業を守る為のインテグリティーとコンプライアンス文化
4.規制、国際標準
品質や規制に無関心なマネジメント層のマインドを変える
全社員への法規制教育は医療機器会社の生命線
専任規制・規格担当者の必要性
FDA QSRとISOの相違とは、ただ差分を理解することではない
5.プロジェクト推進
トップマネジメントが果たす大きな役割(リーダーシップ)
会社の現状を把握し、会社を変革しないとグローバル進出できない
PM(プロダクトマネジャー)とPL(プロダクトリーダー)がコンカレント開発を実現する
FDA設計管理ガイダンスコース Rev.13
〜 規制だけではなく会社に利益をもたらすものづくりの本質 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
満席のためお申込を締め切らせて戴きました。
次回は11月24日開催です!
☆ 開催概要 ☆
ほとんどの日本企業に於ける開発、設計管理の仕組みがFDAが要求する規制に準拠していません。これはグローバルに進出しようとする企業だけではなく、患者の命を預かる全ての医療機器メーカーにとって大きなリスクを伴います。FDAは自らの査察で医療機器品質不良の根本原因を統計的に設計管理にあると見抜いており、それはクオリス・イノーバの多くのGAP監査経験でも明かです。そこで本コースでは、グローバルな開発・設計基準を「FDA設計管理ガイダンス」と、欧米で取り入れられているフレームワークを元に体系的に解説し、医療機器開発の本質である品質不良を発生させない仕組み(ロバスト設計)をタグチメソッドを用いて解説します。ただ規制だからではなく、設計品質を高める為に。エンジニアの方から多くの共感と支持を集めてきたクオリス・イノーバの最も人気のある価値あるコースです。 オンサイトでも可能です。お問い合わせはこちらから。
日 時:2015年6月15日(月曜) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している企業
FDA査察準備を考えている企業
受講料: ¥49,000( テキスト・規制邦訳文・昼食代含む )
割 引: ¥39,000( テキスト・規制邦訳文・昼食代含む )
1社2名以上ご参加の場合、或いはQSRコース(6月1日、2日)を
受講された方
特 割: オンサイトセミナーの後、継続研修などでご利用の場合、特別割引を適用させて頂
きますのでご相談下さい。 この場合、正規料金をお支払い頂く方が優先され、
人気コースの為ご出席戴けない場合がございますので予めご了解下さい。
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
このコースを受講される前にQSR2日コースを受講されることをお勧め致します。
1.設計品質の本質的価値
繰り返される設計品質に起因する製品不良
ISOとQSRの本質的な違い
理解すべき規制・規格・ガイダンス
FDAが望んでいるTQMの本質、品質工学に基づくロバスト設計
医療機器の設計手法( Water Fall Model )
一貫性を問うFDA査察
〜 TQM、FDA設計管理ガイダンスに基づくQSRサブパートCの実践 〜
2.820.30 ( a ) 総則
TQMに欠かせないマネジメントチームの重要な役割
設計環境、開発リソースの見極め
要員教育の重要性( 820.25 )
力量表と教育計画
3.820.30 ( b ) 設計及び開発計画
医療機器開発ライフサイクルマネジメント
リスクマネジメントライフサイクル
タグチメソッドの開発方式によるフロントローディング手法
PM,PLの役割
カスタマーニーズを要求仕様に変換する品質機能展開
設計品質確立の重要な要素、コンカレントエンジニアリング
設計計画書とは
4.820.30 ( c ) 設計インプット
ロバスト設計( タグチメソッド )による未然防止設計
要求仕様書から設計仕様書への変換
曖昧なニーズのスペックへの変換
重要な設計要素トレーサビリティーマトリックスの使用
ラベリング、梱包の設計インプット
5.820.30 ( d ) 設計アウトプット
入力のバラツキを押さえたアウトプットの確認
設計アウトプットを定義し文書類を明確にする
E-BOM / M-BOM とは
製造仕様書とは
図面発行
DMRとのトレーサビリティー
6.820.30 ( j ) 設計履歴ファイル( DHF )
DHF / DMR / DHR の定義を理解する
識別、ファイルの基本
文書体系と設計基準
7.820.30 ( e ) 設計審査
問題の早期発見、解決が目的で、セレモニーで決して終わらない
設計審査で設計のロバストネスを確認
設計審査とテクニカルレビュー
キーとなる審査者
8.820.30 ( f ) 設計検証
設計検証と設計バリデーションの相違
インプットからアウトプットまでトレーサブルであるとは
エビデンスに基づく設計
設計検証レポートは、設計者の活動記録
9.820.30 ( g ) 設計バリデーション
設計バリデーションの手法
社内、臨床評価の相違と記録
ソフトウェアバリデーションとVモデル
3種類のバリデーションの相違
10.820.30 ( h ) 設計移管
設計部門が製造工程を設計
工程V&VとQC工程表
機能展開とPFMEA
製造仕様書への展開
DHF,DMR,DHR
11.820.30 ( i ) 設計変更
重要な査察ポイント
設計変更プロセスとリスクアセスメント
コミュニケーションエラー
製品不良を再発させない重要な要素、設計CAPA
文書、図面の変更管理
( 内容が変更になる場合もございますが大きな変更はございません。 )
2014_12

FDA GMP( QSR )2日間コース Rev.14
〜 医療機器・IVDに求められる規制要求事項の本質を学ぶ実践コース 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
☆ 開催概要 ☆
* 今回からFDA査察対応ネイティブ通訳を含むエキスパートチームで
完訳した対訳本をテキストとは別に提供します。
本コースは、この新たな訳に沿って新しいテキストにしました。
定員オーバーにつき、募集を締め切らせて戴きました!
次回は11月開催です。
グローバルに進出しようとする企業にとって 米国は最も大きなマーケットであり、米国市場での成功がグローバルを制するといっても過言ではありません。しかしながら、米国に上市するには医療機器規制QSRに基づくFDAの査察は避けられません。クオリス・イノーバのQSRコースは、多くのFDA査察準備、対応の経験や品質管理の経験から、FDA規制のみならず医療機器開発・製造の本質を鋭く説き、多くの企業の皆様の共感を集め支持されて参りました。米国に進出する企業のみならず、医療機器・IVD を開発しようとしている企業の皆様のマインドが大きく変わる価値あるクオリス・イノーバのメインコースです。 オンサイトでも可能です。お問い合わせはこちらから。
日 時:2015年6月1日(月)、2日(火) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
受講料: ¥94,000( 2日分、テキスト・対訳本・昼食代含む )
割 引: ¥79,000( 2日割引適用、テキスト・対訳本・昼食代含む )
1社2名様以上ご参加の場合
特 割: 企業で行うオンサイトセミナー開催の後、継続研修などでご利用の場合、
特別割引を適用させて頂きますのでメールでご相談下さい。
この場合、正規料金をお支払いの方が優先となりご希望に添えない場合が
ございますので予めご了承ください。
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
第1日目 セミナー内容
A.FDA規制、査察の最新情報
1.FDA規制、医療機器規制の本質( 医療機器開発・製造の本質を理解する )
2.FDA査察の本質、TQMを理解する
3.なぜISOではFDA査察にパスしないのか
4.ISOマインドを変える
5.根拠に基づくエビデンスの重要性
6,QSITアプローチとは
7.医療機器規制を正しく理解する為に
8.QSR規制要求事項の本質を理解する
820.20 経営者の責任
品質目標展開と品質計画
経営者がリードするTQMを理解すると見えてくる
マネジメントレビューの本質
FDA が一番確認したいこととは。。
820.22 品質監査
効果的な監査手法と監査報告のあり方
内部監査員のFDAエキスパートの相違
820.25 要員
教育の重要性:教育の手法と記録、力量表、リソースとは
アウェアネストレーニングの実施
820.30 設計管理( 概要のみ:設計監理1日コースで詳細解説 )
ライフサイクル製品開発手法、ウオーターフォールモデル、
フロントローディング手法、ロバスト設計
設計検証と設計バリデーションの相違
ISO にない設計移管、工程設計とは
設計CAPAとは
820.40 文書管理
グロバールに通用する文書管理手法と教育
820.50 購買管理
これからの査察の潮流、サプライヤー管理とは
購買チームが品質に貢献するために
820.60 識別
混同を防止する為の識別方法とは
820.65 トレーサビリティー
体内埋込型機器だけではなく、変化点を知り品質を高める
820.80 受領、工程内及び完成機器のアクセプタンス
アクセプタンスを理解する
Receiving, In-process, and Finished device acceptance の相違
820.86 アクセプタンス状態
状態表示する意味
820.90 不適合品
不適合品の処理プロセス
FDAはなぜNCを見たがるのか
グローバル用語を使用する
820.120 機器のラベリング
リコールが一番多く、査察で注目されるラベリングの管理とは
ラベルとラベリングの相違
820.130 機器の梱包
梱包箱を見ればどんな設計を行っていたかが判る
820.140 取扱い
混同・劣化・損傷・汚染を発生させないためのリスク管理
820.150 保管
倉庫などの保管エリアのリスク管理
リスクアセスメントをすると。。
820.160 流通
劣化品の流通を防ぐ設計品質とは
820.170 据付け
インスタレーション手順書のV&V
820.180 記録
全ての活動が根拠に基づくエビデンスで保証されるための重要な活動
Objective Evidence を示すための記録管理とは
820.181 機器原簿(DMR)
DMRを定義し識別する
DHF,DMR,DHRの相違
820.184 機器履歴簿(DHR)
製造記録の管理方法、特に重要な。。
820.186 品質システム記録
DHF,DMR,DHR以外の品質記録とは
820.200 付帯サービス
サービス記録の分析とMR,MDRの判定
820.250 統計的手法
TQMを進めていく上で最も重要な要素
品質改善に欠かせない統計的手法
第2日目 セミナー内容
820.70 生産及び工程管理
工程を監視、管理する工程パラメータとは
工程設計と工程V&V
工程のリスクマネジメント(PFMEA)
要員、環境、汚染管理
設備の妥当性とは
保守と検査の相違
副資材の定義と管理
820.72 検査・測定及び試験装置
意図した用途への適合性、測定器の精度管理とは
測定器の精度管理と救済処置
820.75 工程の妥当性確認
QSR,医療機器規制の本質、工程バリデーションとは
TQMが目指すべき試験レスとは
バリデーションの手法( VMP,DQ、IQ、OQ、PQ、PPQ )
設備バリデーションとは
CSV( コンピュータシステムバリデーション )とは
820.198 苦情ファイル
査察のキーポイント、苦情管理の目的を理解する
苦情処理とMDR( Part 803 )、CAR( Part 806 )
820.100 是正処置及び予防処置( CAPA )
FDA警告書で最も多い指摘CAPAの原則を理解する
修正、是正、予防の相違を正確に理解しISOマインドを変える
CAPA7ステップを理解する
CAPAの効果(再発防止)を出す仕組み
MRで監視すべきCAPAプロセス
( 内容が変更になる場合もございますが大きな変更はございません。)

医療機器の品質問題を防ぐ仕組みと開発手法
〜 ISOの正しい解釈を医療機器規制と事故事例に学ぶ 〜
このコースは【 日経ものづくり 】主催で開催されます。
http://techon.nikkeibp.co.jp/article/SEMINAR/20150202/402050/
終了しました
☆ 開催概要 ☆
日 時: 2015年4月13日(月曜) 10:00 〜 17:00
会 場: Learning Square 新橋 6F( 東京・新橋 )
主 催: 日経ものづくり
お申込: 主催者HPより直接お申し込み下さい
http://techon.nikkeibp.co.jp/article/SEMINAR/20150202/402050/
セミナー内容:
1.なぜ医療機器の品質問題がなくならないのか
「あってはならない」とされる医療機器の品質問題だが、実際にはなかなか減っていない。食品医薬品局(FDA)の査察が厳しい米国では、名だたる大メーカーがリコールに追われている。メーカーも手を打っているはずなのに、なぜ医療機器の品質問題がなくならないのか、一般的な機器の事故事例を基に解説する。
2.品質問題の直接原因と根本原因を区別する
品質問題がなかなか減らない理由の1つに、根本原因までさかのぼって分析していないことが挙げられる。多くの場合、直接原因の特定だけで満足してしまっているため、同じような品質問題を繰り返すことになるのだ。直接原因と根本原因の違いを明らかにした上で、根本原因を見つけるための考え方を紹介する。
3.CAPAとは何か
品質問題を根絶するには、開発体制の問題点を完全に取り除いて品質問題の発生を未然に防ぐCAPA( Corrective Action & Preventive Action、是正処置及び予防処置 )の取組みが欠かせない。しかし、多くの企業では単なる直接原因の"モグラ叩き”に終始している。CAPAの真髄を解説する。
4.製品開発にCAPAを活かす
CAPAのアプローチを自社の製品開発に導入するには、どうすればよいのか。当然、自社のやり方を見直さなければならない部分が出てくる。医療機器メーカー、部品・材料メーカーなどそれぞれの立場に向けて、CAPAを導入する上で基本となる考え方を紹介する。
5.品質問題の事例に学ぶ
直接原因と根本原因の違いを正確に区別し、根本原因までだかのぼって分析できるようになるには、過去の品質問題に学ぶ事が有効である。医療機器に限らずさまざまな事例を題材として取り上げることで、本質を見分けるための"目”を養う。ここでは、『日経ものづくり』のコラム「事故は語る」や、同コラムをまとめた書籍『事故の事典』も活用する。
2015_4_13

FDA プロセスバリデーションコース Rev.9.5
〜 プロセスバリデーションの本質の理解と工程設計 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
定員オーバーの為、募集を締め切らせて頂きました。
☆ 開催概要 ☆
クオリス・イノーバがこれまで実施してきたGAP監査では、プロセスバリデーションのエビデンスが提出できず、手順さえもない企業がほとんどです。エビデンスを提出できたとしてもプロセスバリデーションの本質を理解していないので、監査に耐えられる満足のいくものではありません。品質システムが目指すものは、意図した用途に適合する製品を終始一貫して生産すること。これらの原則と目的を達成するための重要な要素がプロセスバリデーションです。そこで、規制当局はプロセスバリデーションの手順と結果を確認しようとします。FDA査察の結果を統計的にみてもワースト10に入る指摘項目となっています。このコースではISOにはない設計移管から工程設計、マスタープランからCSVを含むプロセスバリデーションを解説、製品品質のバラツキを無くして品質コストを下げ、医療機器の製品安全性に貢献するプロセスバリデーションの本質を理解します。
日 時:2015年3月9日(月曜) 10:30 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している企業
FDA査察準備を考えている企業
受講料: ¥48,000
割 引: ¥38,500
1社2名以上ご参加の場合
定 員: 24名 (3月3日締切)
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
![]()
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
1.なぜ工程バリデーションを実施するのか?
プロセスバリデーションの目的と本質
工程管理の目的と本質
医療機器規制・規格を理解する
2.バリデーションすべき工程とは
バリデーションすべき工程の識別
検証工程とバリデーション工程の違い
設備バリデーションと工程バリデーション
バリデーション工程の例
3.バリデーションの種類
バリデーションの種類
設備バリデーションと劣化パラメータ
コンピュータシステムバリデーション(CSV)
4.バリデーションステップ
VMP: バリデーションマスタープラン
DQ: 設計時的確性評価
IQ: 据付時的確性評価
OQ: 稼働時的確性評価
PQ: 製造性能的確性評価
バリデーションの文書化
プロセスバリデーションの明確化
自動化工程の明確化
CSVカテゴリーの明確化
ソフトウェアバリデーションの基本
カテゴリー別ソフトウェア
5.設計移管とプロセスバリデーション
設計移管とプロセスバリデーションの関係
ミニスケールとフルスケール
工程設計表とトレーサビリティー
6.バリデーションの実践
マスターバリデーションプラン
バリデーション工程の識別
バリデーション計画
設備リストとバリデーション工程リスト
チームビルディング
DQ Plan /DQ Report
IQ Plan/IQ Report
OQ Plan/OQ Report
PQ Plan/PQ Report
7.バリデーションのアウトプット
QC工程表と管理図
工程能力
再バリデーションの時期設定
設備メンテナンス手順
*内容は変更される場合がございます*
昨年度講習風景
CAPA コース Rev.14
〜 査察で最も指摘の多いCAPAの正しい理解と問題を再発させない仕組み 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
終了致しました
☆ 開催概要 ☆
FDA査察の結果を統計的にまとめてみるとCAPAで最も多くの指摘をしていることが判ります。これは日本企業の指摘でも顕著です。FDAは自らの査察を通して企業で最も理解されていないプロセスを統計的によく理解しており、不適合の再発原因がCAPAにあることを知っているからです。このことはクオリス・イノーバが実施してきたGAP監査でも明かで、日本のほとんどの企業でCAPAを正しく理解しておらずマネジメントできていません。また、FDAの査察を受けた後のアクションレターもCAPAのステップで回答すべきです。このコースに参加指されたほとんどの方のCAPAのマインドが変わる価値あるコースで、医療機器だけではなく、医薬品や多くの業種で理解すべき内容です。
* 今回の Rev.14 では Rev.13 に加え、理解を助ける為、事例を多く取り上げています。
日 時:2015年3月2日(月曜) 10:30 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVDに限らず医薬品などCAPAを用いた品質改善を期待される企業
受講料: ¥48,000( テキスト・昼食代含む )
割 引: ¥38,500( テキスト・昼食代含む )
1社2名以上ご参加の場合
特 割: オンサイトセミナーの後、継続研修などでご利用の場合、特別割引を適用させて
頂きますのでご相談下さい。 尚、この場合、正規料金をお支払い頂く方が優先
され出席戴けない場合がございますので予めご了解下さい。
定 員: 20名 2月24日締切
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
![]()
☆ セミナー内容 ☆
1.なぜ事故は再発するのか?
2.会社の文化、価値観が重要な要素
3.規制・ISO規格を理解する
4.ガイドラインの理解
5.TQMの理解と仕組みが無いとCAPAは回らない
7.再発する品質不良
8.修正・是正・予防の正しい理解
修正・CA・PAの相違
9.医療機器規制要求事項
FDA 21CFR Part 820.100 CAPA とISO 13485 8.5 改善
10.不適合、逸脱、特別採用と逸脱許可の相違
11.修正、手直し、修理の相違
12.苦情処理、不適合処理、CAPA手順との関係
13.修正プロセス
14.具体的なCA、PAエスカレーション
15.CAPA7ステップ
Six Sigma との相違
Step 1 問題を検出し明確にする
Step 2 問題の原因を詳しく調査する
Step 3 根本原因を特定する
Step 4 是正又は予防するための処置
Step 5 処置の結果を文書化
Step 6 処置の検証・妥当性確認
Step 7 有効性の確認
16.CAPAのマネジメント
17.設計CAPA
18.Warning Letter Case Study
19.ケーススタディ
*内容は変更される場合がございます*
昨年度講習風景
FDA設計管理ガイダンスコース Rev.13
〜 規制だけではなく会社に利益をもたらすものづくりの本質 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
☆ 開催概要 ☆
定員オーバーの為、募集を終了させて戴きました。
次回は来年5月以降となります!
ほとんどの日本企業に於ける開発、設計管理の仕組みがFDAが要求する規制に準拠していません。これはグローバルに進出しようとする企業だけではなく、患者の命を預かる全ての医療機器メーカーにとって大きなリスクを伴います。FDAは自らの査察で医療機器品質不良の根本原因を統計的に設計管理にあると見抜いており、それはクオリス・イノーバの多くのGAP監査経験でも明かです。そこで本コースでは、グローバルな開発・設計基準を「FDA設計管理ガイダンス」と、欧米で取り入れられているフレームワークを元に体系的に解説し、医療機器開発の本質である品質不良を発生させない仕組み(ロバスト設計)をタグチメソッドを用いて解説します。ただ規制だからではなく、設計品質を高める為に。エンジニアの方から多くの共感と支持を集めてきたクオリス・イノーバの最も人気のある価値あるコースです。 オンサイトでも可能です。お問い合わせはこちらから。
日 時:2014年12月8日(月曜) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している企業
FDA査察準備を考えている企業
受講料: ¥49,000( テキスト・規制邦訳文・昼食代含む )
割 引: ¥39,000( テキスト・規制邦訳文・昼食代含む )
1社2名以上ご参加の場合、或いはQSRコース(12月1日、2日)を
受講された方
特 割: オンサイトセミナーの後、継続研修などでご利用の場合、特別割引を適用させて頂
きますのでご相談下さい。 この場合、正規料金をお支払い頂く方が優先され、
人気コースの為ご出席戴けない場合がございますので予めご了解下さい。
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
このコースを受講される前にQSR2日コースを受講されることをお勧め致します。
1.設計品質の本質的価値
繰り返される設計品質に起因する製品不良
ISOとQSRの本質的な違い
理解すべき規制・規格・ガイダンス
FDAが望んでいるTQMの本質、品質工学に基づくロバスト設計
医療機器の設計手法( Water Fall Model )
一貫性を問うFDA査察
〜 TQM、FDA設計管理ガイダンスに基づくQSRサブパートCの実践 〜
2.820.30 ( a ) 総則
TQMに欠かせないマネジメントチームの重要な役割
設計環境、開発リソースの見極め
要員教育の重要性( 820.25 )
力量表と教育計画
3.820.30 ( b ) 設計及び開発計画
医療機器開発ライフサイクルマネジメント
リスクマネジメントライフサイクル
タグチメソッドの開発方式によるフロントローディング手法
PM,PLの役割
カスタマーニーズを要求仕様に変換する品質機能展開
設計品質確立の重要な要素、コンカレントエンジニアリング
設計計画書とは
4.820.30 ( c ) 設計インプット
ロバスト設計( タグチメソッド )による未然防止設計
要求仕様書から設計仕様書への変換
曖昧なニーズのスペックへの変換
重要な設計要素トレーサビリティーマトリックスの使用
ラベリング、梱包の設計インプット
5.820.30 ( d ) 設計アウトプット
入力のバラツキを押さえたアウトプットの確認
設計アウトプットを定義し文書類を明確にする
E-BOM / M-BOM とは
製造仕様書とは
図面発行
DMRとのトレーサビリティー
6.820.30 ( j ) 設計履歴ファイル( DHF )
DHF / DMR / DHR の定義を理解する
識別、ファイルの基本
文書体系と設計基準
7.820.30 ( e ) 設計審査
問題の早期発見、解決が目的で、セレモニーで決して終わらない
設計審査で設計のロバストネスを確認
設計審査とテクニカルレビュー
キーとなる審査者
8.820.30 ( f ) 設計検証
設計検証と設計バリデーションの相違
インプットからアウトプットまでトレーサブルであるとは
エビデンスに基づく設計
設計検証レポートは、設計者の活動記録
9.820.30 ( g ) 設計バリデーション
設計バリデーションの手法
社内、臨床評価の相違と記録
ソフトウェアバリデーションとVモデル
3種類のバリデーションの相違
10.820.30 ( h ) 設計移管
設計部門が製造工程を設計
工程V&VとQC工程表
機能展開とPFMEA
製造仕様書への展開
DHF,DMR,DHR
11.820.30 ( i ) 設計変更
重要な査察ポイント
設計変更プロセスとリスクアセスメント
コミュニケーションエラー
製品不良を再発させない重要な要素、設計CAPA
文書、図面の変更管理
( 内容が変更になる場合もございますが大きな変更はございません。 )

FDA GMP( QSR )2日間コース Rev.13
〜 医療機器・IVDに求められる規制要求事項の本質を学ぶ実践コース 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
☆ 開催概要 ☆
定員オーバーの為、募集を終了させて戴きました。
次回は来年5月以降となります!
グローバルに進出しようとする企業にとって 米国は最も大きなマーケットであり、米国市場での成功がグローバルを制するといっても過言ではありません。しかし、米国に上市するには医療機器規制QSRに基づくFDAの査察は避けられません。クオリス・イノーバのQSRコースは、多くのFDA査察準備、対応の経験や品質管理の経験から、FDA規制のみならず医療機器開発・製造の本質を鋭く説き、多くの企業の皆様の共感を集め支持されて参りました。米国に進出する企業のみならず、医療機器・IVD を開発しようとしている企業の皆様のマインドが大きく変わる価値あるクオリス・イノーバのメインコースです。 オンサイトでも可能です。お問い合わせはこちらから。
日 時:2014年12月1日(月)、2日(火) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
受講料: ¥92,000( 2日割引適用、テキスト・規制邦訳文・昼食代含む )
割 引: ¥78,000( 2日分、テキスト・規制邦訳文・昼食代含む )
1社2名以上ご参加の場合
特 割: 企業で行うオンサイトセミナー開催の後、継続研修などでご利用の場合、
特別割引を適用させて頂きますのでメールでご相談下さい。
この場合、正規料金をお支払いの方が優先となりご希望に添えない場合が
ございますので予めご了承ください。
定 員: 24名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
第1日目 セミナー内容
A.FDA規制、査察の最新情報
1.FDA規制、医療機器規制の本質( 医療機器開発・製造の本質を理解する )
2.FDA査察の本質、TQMを理解する
3.なぜISOではFDA査察にパスしないのか
4.ISOマインドを変える
5.根拠に基づくエビデンスの重要性
6,QSITアプローチとは
7.医療機器規制を正しく理解する為に
8.QSR規制要求事項の本質を理解する
820.20 経営者の責任
品質目標展開と品質計画
経営者がリードするTQMを理解すると見えてくるマネジメントレビューの本質
FDA が一番確認したいこととは。。
820.22 品質監査
効果的な監査手法と監査報告のあり方
内部監査員のFDAエキスパートの相違
820.25 要員
教育の重要性:教育の手法と記録、力量表、リソースとは
アウェアネストレーニングの実施
820.30 設計管理( 概要のみ:設計監理1日コースで詳細解説 )
ライフサイクル製品開発手法、ウオーターフォールモデル、
フロントローディング手法、ロバスト設計
設計検証と設計バリデーションの相違
ISO にない設計移管、工程設計とは
設計CAPAとは
820.40 文書管理
グロバールに通用する文書管理手法と教育
820.50 購買管理
これからの査察の潮流、サプライヤー管理とは
購買チームが品質に貢献するために
820.60 識別
混同を防止する為の識別方法とは
820.65 トレーサビリティー
体内埋込型機器だけではなく、変化点を知り品質を高める
820.80 受領、工程内及び完成品の受入
Receiving, In-process, and Finished device acceptance の相違
820.86 受入の状態
状態表示する意味
820.90 不適合品
不適合品の処理プロセス
FDAはなぜNCを見たがるのか
グローバル用語を使用する
820.120 機器のラベリング
リコールが一番多く、査察で注目されるラベリングの管理とは
ラベルとラベリングの相違
820.130 機器の梱包
梱包箱を見ればどんな設計を行っていたかが判る
820.140 取り扱い
混同・劣化・損傷・汚染を発生させないためのリスク管理
820.150 保管
倉庫などの保管エリアのリスク管理
リスクアセスメントをすると。。
820.160 流通
劣化品の流通を防ぐ設計品質とは
820.170 据付
インスタレーション手順書のV&V
820.180 記録
全ての活動が根拠に基づくエビデンスで保証されるための重要な活動
Objective Evidence を示すための記録管理とは
820.181 機器原簿(DMR)
DMRを定義し識別する
DHF,DMR,DHRの相違
820.184 機器履歴簿(DHR)
製造記録の管理方法、特に重要な。。
820.186 品質システム記録
DHF,DMR,DHR以外の品質記録とは
820.200 付帯サービス
サービス記録の分析とMR,MDRの判定
820.250 統計的手法
TQMを進めていく上で最も重要な要素
品質改善に欠かせない統計的手法
第2日目 セミナー内容
820.70 生産及び工程管理
工程を監視、管理する工程パラメータとは
工程設計と工程V&V
工程のリスクマネジメント(PFMEA)
要員、環境、汚染管理
設備の妥当性とは
保守と検査の相違
副資材の定義と管理
820.72 検査・測定及び試験装置
意図した用途への適合性、測定器の精度管理とは
測定器の精度管理と救済処置
820.75 工程の妥当性確認
QSR,医療機器規制の本質、工程バリデーションとは
TQMが目指すべき試験レスとは
バリデーションの手法( VMP,DQ、IQ、OQ、PQ、PPQ )
設備バリデーションとは
CSV( コンピュータシステムバリデーション )とは
820.198 苦情ファイル
査察のキーポイント、苦情管理の目的を理解する
苦情処理とMDR( Part 803 )、CAR( Part 806 )
820.100 是正処置及び予防処置( CAPA )
FDA警告書で最も多い指摘CAPAの原則を理解する
修正、是正、予防の相違を正確に理解しISOマインドを変える
CAPA7ステップを理解する
CAPAの効果(再発防止)を出す仕組み
MRで監視すべきCAPAプロセス
( 内容が変更になる場合もございますが大きな変更はございません。)

FDA QSR 内部監査員コース
〜 QSR 対応内部監査員養成のための社内資格認定コース 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
満席になりましたので募集を締め切らせて戴きました。
次回セミナーでお待ちしております!
☆ 開催概要 ☆
不適合を出さないようプロセスに内在する不適合を未然に抽出し、マネジメントレビューでトップマネジメントの指示のもとプロセス改善を常に行う。これがTQMの有るべき姿であり、規制当局が求めている姿です。一方現実は、プロセスを監視し傾向管理を行っておらず、よく訓練されていないにわか監査員がISOで要求されているからといって片手間に監査を行っているだけ。これは医療機器の場合致命的で、リコールで何十億の損失を出すだけではなく、人の命に関わると会社の存続まで危ぶまれます。このコースではISO内部監査員の資格を持つ方々がQSRの内部監査が行えるよう社内資格を付与するための社内資格認定コースです。一般に行われているISOの内部監査手法とは異なる、付加価値を生み、予防対策としての監査手法とノウハウを学びます。
日 時:2014年10月10日(金曜) 10:00 〜 16:00
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:すでに弊社主催下記コースをオープン、オンサイト問わず受講された方限定です。
* FDA GMP QSR 2日間コース
* FDA 設計管理コース
このコースはQSRの基礎知識が無いと受講できません。弊社のコースを受講されていない
場合、内容の理解が進まず、異なる解釈による質問でコースの進捗を妨げてしまう為です。
何卒ご理解をお願い申し上げます。
受講料: ¥49,000( テキスト、QSIT邦訳、昼食代含む、内税 )
このコースは割引がございません。
定 員: 20名 満席になり次第締切ます
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
過去に受講された弊社QSR2日間コースと設計管理コースの受講日を記載して下さい。
オンサイトで受講された場合でも構いません。
折り返し、会場、お振込先情報をメールでご連絡致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
1.なぜ内部監査を行うのか?
規制・規格が求める目的を理解する
患者の命を優先する監査とは?
企業の付加価値を高める為には?
2.内部監査員に求められる力量
内部監査員とリーダーに求められる力量
内部監査員トレーニング
マネジメントチームのカウンセラー
3.プロセスアプローチ
プロセスのR&Rを明確にする
プロセスを常に測定し改善する
内部監査はプロセスに注目
4.監査判定基準
チェックリストと数値化
プロセス毎に数値化
FDA査察と内部監査判断基準の相違を知る
5.監査を計画する
マネジメントレビューと品質会議
プロセス管理パラメートと傾向分析結果
是正監査と予防監査の相違
監査は短期集中型で実施
監査員レビュー会のノウハウ
6.監査の目のつけどころ( QSIT )
1.経営者による管理
2.設計管理
3.CAPA
MDR報告
CAR
MDトラッキング
4.P&PC
5.滅菌工程
*内容は変更される場合がございます*
前回講習風景

FDA プロセスバリデーションコース Rev.9
〜 プロセスバリデーションの本質の理解と製品品質向上 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
満席になりましたので募集を締め切らせて戴きました
次回セミナーでお待ちしております!
☆ 開催概要 ☆
クオリス・イノーバがこれまで実施してきたGAP監査では、プロセスバリデーションのエビデンスが提出できず、手順さえもない企業がほとんどです。エビデンスを提出できたとしてもプロセスバリデーションの本質を理解していないので、監査に耐えられる満足のいくものではありません。品質システムが目指すものは、意図した用途に適合する製品を終始一貫して生産すること。これらの原則と目的を達成するための重要な要素がプロセスバリデーションです。そこで、規制当局はプロセスバリデーションの手順と結果を確認しようとします。FDA査察の結果を統計的にみてもワースト10に入る指摘項目となっています。このコースではISOにはない設計移管から工程設計、マスタープランからCSVを含むプロセスバリデーションを解説、製品品質のバラツキを無くして品質コストを下げ、医療機器の製品安全性に貢献するプロセスバリデーションの本質を理解します。
日 時:2014年7月14日(月曜) 10:30 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している企業
FDA査察準備を考えている企業
受講料: ¥48,000
割 引: ¥38,500
1社2名以上ご参加の場合、或いは
QSRコース(6月2、3日)、設計監理コース(6月9日)、CAPAコース
(6月23日)を受講された方。
定 員: 24名 満席になり次第締切ます( 30名に変更させて戴きました )
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
1.なぜ工程バリデーションを実施するのか?
プロセスバリデーションの目的と本質
工程管理の目的と本質
医療機器規制・規格を理解する
2.バリデーションすべき工程とは
バリデーションすべき工程の識別
検証工程とバリデーション工程の違い
設備バリデーションと工程バリデーション
バリデーション工程の例
3.バリデーションの種類
バリデーションの種類
設備バリデーションと劣化パラメータ
コンピュータシステムバリデーション(CSV)
4.バリデーションステップ
VMP: バリデーションマスタープラン
DQ: 設計時的確性評価
IQ: 据付時的確性評価
OQ: 稼働時的確性評価
PQ: 製造性能的確性評価
バリデーションの文書化
プロセスバリデーションの明確化
自動化工程の明確化
CSVカテゴリーの明確化
ソフトウェアバリデーションの基本
カテゴリー別ソフトウェア
5.設計移管とプロセスバリデーション
設計移管とプロセスバリデーションの関係
ミニスケールとフルスケール
工程設計表とトレーサビリティー
6.バリデーションの実践
マスターバリデーションプラン
バリデーション工程の識別
バリデーション計画
設備リストとバリデーション工程リスト
チームビルディング
DQ Plan /DQ Report
IQ Plan/IQ Report
OQ Plan/OQ Report
PQ Plan/PQ Report
7.バリデーションのアウトプット
QC工程表と管理図
工程能力
再バリデーションの時期設定
設備メンテナンス手順
*内容は変更される場合がございます*
昨年度講習風景

CAPA コース Rev.13
〜 査察で最も指摘の多いCAPAの正しい理解と問題を再発させない仕組み 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
募集を締め切らせて戴きました
☆ 開催概要 ☆
FDA査察の結果を統計的にまとめてみるとCAPAで最も多くの指摘をしていることが判ります。これは日本企業の指摘でも顕著です。FDAは自らの査察を通して企業で最も理解されていないプロセスを統計的によく理解しており、不適合の再発原因がCAPAにあることを知っているからです。このことはクオリス・イノーバが実施してきたGAP監査でも明かで、日本のほとんどの企業でCAPAを正しく理解しておらずマネジメントできていません。また、FDAの査察を受けた後のアクションレターもCAPAのステップで回答すべきです。このコースに参加指されたほとんどの方のCAPAのマインドが変わる価値あるコースで、医療機器だけではなく、医薬品や多くの業種で理解すべき内容です。
日 時:2014年6月23日(月曜) 10:30 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVDに限らず医薬品など広くCAPAを用いた品質改善を期待される企業
受講料: ¥48,000( テキスト・昼食代含む )
割 引: ¥38,500( テキスト・昼食代含む )
1社2名以上ご参加の場合、或いは
QSRコース(6月2、3日)、設計監理コース(6月9日)を受講された方
特 割: すでに多くの方が受講されている企業様、オンサイトセミナーの後、継続研修などで
ご利用の場合、特別割引を適用させて頂きますのでご相談下さい。
正規料金をお支払い頂く方が優先され出席戴けない場合がございますので予めご了解
下さい。
定 員: 20名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
1.なぜ事故は再発するのか?
2.会社の文化、価値観が重要な要素
3.規制・ISO規格を理解する
4.ガイドラインの理解
5.TQMの理解と仕組みが無いとCAPAは回らない
7.再発する品質不良
8.修正・是正・予防の正しい理解
修正・CA・PAの相違
9.医療機器規制要求事項
FDA 21CFR Part 820.100 CAPA とISO 13485 8.5 改善
10.不適合、逸脱、特別採用と逸脱許可の相違
11.修正、手直し、修理の相違
12.苦情処理、不適合処理、CAPA手順との関係
13.修正プロセス
14.具体的なCA、PAエスカレーション
15.CAPA7ステップ
Six Sigma との相違
Step 1 問題を検出し明確にする
Step 2 問題の原因を詳しく調査する
Step 3 根本原因を特定する
Step 4 是正又は予防するための処置
Step 5 処置の結果を文書化
Step 6 処置の検証・妥当性確認
Step 7 有効性の確認
16.CAPAのマネジメント
17.設計CAPA
18.Warning Letter Case Study
*内容は変更される場合がございます*
昨年度講習風景

FDA設計管理ガイダンスコース Rev.12
〜 規制だけではなく会社に利益をもたらすものづくりの本質 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
募集を締め切らせて戴きました。
次回開催は12月8日です。
☆ 開催概要 ☆
ほとんどの日本企業に於ける開発、設計管理の仕組みがFDAが要求する規制に準拠していません。これはグローバルに進出しようとする企業だけではなく、患者の命を預かる全ての医療機器メーカーにとって大きなリスクを伴います。FDAは自らの査察で医療機器品質不良の根本原因を統計的に設計管理にあると見抜いており、それはクオリス・イノーバの多くのGAP監査経験でも明かです。そこで本コースでは、グローバルな開発・設計基準を「FDA設計管理ガイダンス」と、欧米で取り入れられているフレームワークを元に体系的に解説し、医療機器開発の本質である品質不良を発生させない仕組み(ロバスト設計)をタグチメソッドを用いて解説します。ただ規制だからではなく、設計品質を高める為に。エンジニアの方から多くの共感と支持を集めてきたクオリス・イノーバの最も人気のある価値あるコースです。
日 時:2014年6月9日(月曜) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にソフトウェア搭載機器でこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している企業
FDA査察準備を考えている企業
受講料: ¥49,000( テキスト・規制邦訳文・昼食代含む )
割 引: ¥39,000( テキスト・規制邦訳文・昼食代含む )
1社2名以上ご参加の場合、或いはQSRコース(6月2日、3日)を受講された方
特 割: オンサイトセミナーの後、継続研修などでご利用の場合、特別割引を適用させて頂き
ますのでご相談下さい。 この場合、正規料金をお支払い頂く方が優先され、人気
コースの為ご出席戴けない場合がございますので予めご了解下さい。
定 員: 30名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
このコースを受講される前にQSR2日コースを受講されることをお勧め致します。
1.設計品質の本質的価値
繰り返される設計品質に起因する製品不良
ISOとQSRの本質的な違い
理解すべき規制・規格・ガイダンス
FDAが望んでいるTQMの本質、品質工学に基づくロバスト設計
医療機器の設計手法( Water Fall Model )
一貫性を問うFDA査察
〜 TQM、FDA設計管理ガイダンスに基づくQSRサブパートCの実践 〜
2.820.30 ( a ) 総則
TQMに欠かせないマネジメントチームの重要な役割
設計環境、開発リソースの見極め
要員教育の重要性( 820.25 )
力量表と教育計画
3.820.30 ( b ) 設計及び開発計画
医療機器開発ライフサイクルマネジメント
リスクマネジメントライフサイクル
タグチメソッドの開発方式によるフロントローディング手法
PM,PLの役割
カスタマーニーズを要求仕様に変換する品質機能展開
設計品質確立の重要な要素、コンカレントエンジニアリング
設計計画書とは
4.820.30 ( c ) 設計インプット
ロバスト設計( タグチメソッド )による未然防止設計
要求仕様書から設計仕様書への変換
曖昧なニーズのスペックへの変換
重要な設計要素トレーサビリティーマトリックスの使用
ラベリング、梱包の設計インプット
5.820.30 ( d ) 設計アウトプット
入力のバラツキを押さえたアウトプットの確認
設計アウトプットを定義し文書類を明確にする
E-BOM / M-BOM とは
製造仕様書とは
図面発行
DMRとのトレーサビリティー
6.820.30 ( j ) 設計履歴ファイル( DHF )
DHF / DMR / DHR の定義を理解する
識別、ファイルの基本
文書体系と設計基準
7.820.30 ( e ) 設計審査
問題の早期発見、解決が目的で、セレモニーで決して終わらない
設計審査で設計のロバストネスを確認
設計審査とテクニカルレビュー
キーとなる審査者
8.820.30 ( f ) 設計検証
設計検証と設計バリデーションの相違
インプットからアウトプットまでトレーサブルであるとは
エビデンスに基づく設計
設計検証レポートは、設計者の活動記録
9.820.30 ( g ) 設計バリデーション
設計バリデーションの手法
社内、臨床評価の相違と記録
ソフトウェアバリデーションとVモデル
3種類のバリデーションの相違
10.820.30 ( h ) 設計移管
設計部門が製造工程を設計
工程V&VとQC工程表
機能展開とPFMEA
製造仕様書への展開
DHF,DMR,DHR
11.820.30 ( i ) 設計変更
重要な査察ポイント
設計変更プロセスとリスクアセスメント
コミュニケーションエラー
製品不良を再発させない重要な要素、設計CAPA
文書、図面の変更管理
( 内容が変更になる場合もございますが大きな変更はございません。 )

FDA GMP( QSR )2日間コース Rev.12
〜 医療機器・IVDに求められる規制要求事項の本質を学ぶ実践コース 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
満席につき募集を締め切らせて頂きました
次回セミナーでお待ちしております!
☆ 開催概要 ☆
グローバルに進出しようとする企業にとって 米国は最も大きなマーケットであり、米国市場での成功がグローバルを制するといっても過言ではありません。しかし、米国に上市するには医療機器規制QSRに基づくFDAの査察は避けられません。クオリス・イノーバのQSRコースは、多くのFDA査察準備、対応の経験や品質管理の経験から、FDA規制のみならず医療機器開発・製造の本質を鋭く説き、多くの企業の皆様の共感を集め支持されて参りました。米国に進出する企業のみならず、医療機器・IVD を開発しようとしている企業の皆様のマインドが大きく変わる価値あるクオリス・イノーバのメインコースです。
日 時:2014年6月2日(月)、3日(火) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
受講料: ¥92,000( 2日割引適用、テキスト・規制邦訳文・昼食代含む )
割 引: ¥78,000( 2日分、テキスト・規制邦訳文・昼食代含む )
1社2名以上ご参加の場合
特 割: 企業で行うオンサイトセミナー開催の後、継続研修などでご利用の場合、
特別割引を適用させて頂きますのでメールでご相談下さい。
この場合、正規料金をお支払いの方が優先となりご希望に添えない場合が
ございますので予めご了承ください。
定 員: 30名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
第1日目 セミナー内容
A.FDA規制、査察の最新情報
1.FDA規制、医療機器規制の本質( 医療機器開発・製造の本質を理解する )
2.FDA査察の本質、TQMを理解する
3.なぜISOではFDA査察にパスしないのか
4.ISOマインドを変える
5.根拠に基づくエビデンスの重要性
6,QSITアプローチとは
7.医療機器規制を正しく理解する為に
8.QSR規制要求事項の本質を理解する
820.20 経営者の責任
品質目標展開と品質計画
経営者がリードするTQMを理解すると見えてくるマネジメントレビューの本質
FDA が一番確認したいこととは。。
820.22 品質監査
効果的な監査手法と監査報告のあり方
内部監査員のFDAエキスパートの相違
820.25 要員
教育の重要性:教育の手法と記録、力量表、リソースとは
アウェアネストレーニングの実施
820.30 設計管理( 概要のみ:設計監理1日コースで詳細解説 )
ライフサイクル製品開発手法、ウオーターフォールモデル、
フロントローディング手法、ロバスト設計
設計検証と設計バリデーションの相違
ISO にない設計移管、工程設計とは
設計CAPAとは
820.40 文書管理
グロバールに通用する文書管理手法と教育
820.50 購買管理
これからの査察の潮流、サプライヤー管理とは
購買チームが品質に貢献するために
820.60 識別
混同を防止する為の識別方法とは
820.65 トレーサビリティー
体内埋込型機器だけではなく、変化点を知り品質を高める
820.80 受領、工程内及び完成品の受入
Receiving, In-process, and Finished device acceptance の相違
820.86 受入の状態
状態表示する意味
820.90 不適合品
不適合品の処理プロセス
FDAはなぜNCを見たがるのか
グローバル用語を使用する
820.120 機器のラベリング
リコールが一番多く、査察で注目されるラベリングの管理とは
ラベルとラベリングの相違
820.130 機器の梱包
梱包箱を見ればどんな設計を行っていたかが判る
820.140 取り扱い
混同・劣化・損傷・汚染を発生させないためのリスク管理
820.150 保管
倉庫などの保管エリアのリスク管理
リスクアセスメントをすると。。
820.160 流通
劣化品の流通を防ぐ設計品質とは
820.170 据付
インスタレーション手順書のV&V
820.180 記録
全ての活動が根拠に基づくエビデンスで保証されるための重要な活動
Objective Evidence を示すための記録管理とは
820.181 機器原簿(DMR)
DMRを定義し識別する
DHF,DMR,DHRの相違
820.184 機器履歴簿(DHR)
製造記録の管理方法、特に重要な。。
820.186 品質システム記録
DHF,DMR,DHR以外の品質記録とは
820.200 付帯サービス
サービス記録の分析とMR,MDRの判定
820.250 統計的手法
TQMを進めていく上で最も重要な要素
品質改善に欠かせない統計的手法
第2日目 セミナー内容
820.70 生産及び工程管理
工程を監視、管理する工程パラメータとは
工程設計と工程V&V
工程のリスクマネジメント(PFMEA)
要員、環境、汚染管理
設備の妥当性とは
保守と検査の相違
副資材の定義と管理
820.72 検査・測定及び試験装置
意図した用途への適合性、測定器の精度管理とは
測定器の精度管理と救済処置
820.75 工程の妥当性確認
QSR,医療機器規制の本質、工程バリデーションとは
TQMが目指すべき試験レスとは
バリデーションの手法( VMP,DQ、IQ、OQ、PQ )
設備バリデーションとは
CSV( コンピュータシステムバリデーション )とは
820.198 苦情ファイル
査察のキーポイント、苦情管理の目的を理解する
苦情処理とMDR( Part 803 )、CAR( Part 806 )
820.100 是正処置及び予防処置( CAPA )
FDA警告書で最も多い指摘CAPAの原則を理解する
修正、是正、予防の相違を正確に理解しISOマインドを変える
CAPA7ステップを理解する
CAPAの効果(再発防止)を出す仕組み
MRで監視すべきCAPAプロセス
( 内容が変更になる場合もございますが大きな変更はございません。)

CAPA コース Rev.12
〜 査察で最も指摘の多いCAPAの正しい理解と問題を再発させない仕組み 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
終了しました!
☆ 開催概要 ☆
FDA査察の結果を統計的にまとめてみるとCAPAで最も多くの指摘をしていることが判ります。これは日本企業の指摘でも顕著です。FDAは自らの査察を通して企業で最も理解されていないプロセスを統計的によく理解しており、不適合の再発原因がCAPAにあることを知っているからです。このことはクオリス・イノーバが実施してきたGAP監査でも明かで、日本のほとんどの企業でCAPAを正しく理解しておらずマネジメントできていません。また、FDAの査察を受けた後のアクションレターもCAPAのステップで回答すべきです。このコースに参加指されたほとんどの方のCAPAのマインドが変わる価値あるコースで、医療機器だけではなく、医薬品や多くの業種で理解すべき内容です。
日 時:2013年11月25日(月曜) 10:30 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVDに限らず医薬品など広くCAPAを用いた品質改善を期待される企業
受講料: ¥45,000( テキスト・昼食代含む )
割 引: ¥35,000( テキスト・昼食代含む )
1社2名以上ご参加の場合、或いは
QSRコース(10月28、29日)、設計監理コース(11月11日)を受講された方
特 割: すでに多くの方が受講されている企業様、オンサイトセミナーの後、継続研修などで
ご利用の場合、特別割引を適用させて頂きますのでご相談下さい。
正規料金をお支払い頂く方が優先され出席戴けない場合がございますので予めご了解
下さい。
定 員: 30名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
1.なぜ事故は再発するのか?
2.会社の文化、価値観が重要な要素
3.規制・ISO規格を理解する
4.ガイドラインの理解
5.TQMの理解と仕組みが無いとCAPAは回らない
7.再発する品質不良
8.修正・是正・予防の正しい理解
修正・CA・PAの相違
9.医療機器規制要求事項
FDA 21CFR Part 820.100 CAPA とISO 13485 8.5 改善
10.不適合、逸脱、特別採用と逸脱許可の相違
11.修正、手直し、修理の相違
12.苦情処理、不適合処理、CAPA手順との関係
13.修正プロセス
14.具体的なCA、PAエスカレーション
15.CAPA7ステップ
Six Sigma との相違
Step 1 問題を検出し明確にする
Step 2 問題の原因を詳しく調査する
Step 3 根本原因を特定する
Step 4 是正又は予防するための処置
Step 5 処置の結果を文書化
Step 6 処置の検証・妥当性確認
Step 7 有効性の確認
16.CAPAのマネジメント
17.設計CAPA
18.Warning Letter Case Study
*内容は変更される場合がございます*
昨年度講習風景

FDA設計管理ガイダンスコース Rev.11
〜 設計管理規制だけではなく会社に利益をもたらすものづくりの本質 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
終了しました!
☆ 開催概要 ☆
日本企業のほとんどの開発、設計管理の仕組みがFDAが要求する規制に準拠していません。これはグローバルに進出しようとする企業だけではなく、患者の命を預かる医療機器メーカーにとって大きなリスクを伴います。FDAは自らの査察で医療機器品質不良の根本原因を統計的に設計管理であると見抜いており、それはクオリス・イノーバの多くのGAP監査経験でも明かです。そこで本コースでは、グローバルな開発・設計基準を「FDA設計管理ガイダンス」と、欧米で取り入れられているフレームワークを元に体系的に解説し、医療機器開発の本質である品質不良を発生させない仕組み(ロバスト設計)をタグチメソッドを用いて解説します。ただ規制だからではなく、設計品質を高める為に。エンジニアの方から多くの共感と支持を集めてきたクオリス・イノーバの最も人気のある価値あるコースです。
日 時:2013年11月11日(月曜) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にソフトウェア搭載機器でこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している企業
FDA査察準備を考えている企業
受講料: ¥48,000( テキスト・規制邦訳文・昼食代含む )
割 引: ¥38,000( テキスト・規制邦訳文・昼食代含む )
1社2名以上ご参加の場合、或いはQSRコース(10月28、29日)を受講された方
特 割: すでに多くの方が受講されている企業様、オンサイトセミナーの後、継続研修などで
ご利用の場合、特別割引を適用させて頂きますのでご相談下さい。
正規料金をお支払い頂く方が優先され、人気コースの為ご出席戴けない場合が
ございますので予めご了解下さい。
定 員: 40名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
1.設計品質の本質的価値
繰り返される設計品質に起因する製品不良
ISOとQSRの本質的な違い
理解すべき規制・規格・ガイダンス
FDAが望んでいるTQMの本質、品質工学に基づくロバスト設計
医療機器の設計手法( Water Fall Model )
一貫性を問うFDA査察
〜 TQM、FDA設計管理ガイダンスに基づくQSRサブパートCの実践 〜
2.820.30 ( a ) 総則
TQMに欠かせないマネジメントチームの重要な役割
設計環境、開発リソースの見極め
要員教育の重要性( 820.25 )
力量表と教育計画
3.820.30 ( b ) 設計及び開発計画
医療機器開発ライフサイクルマネジメント
リスクマネジメントライフサイクル
タグチメソッドの開発方式によるフロントローディング手法
PM,PLの役割
カスタマーニーズを要求仕様に変換する品質機能展開
設計品質確立の重要な要素、コンカレントエンジニアリング
設計計画書とは
4.820.30 ( c ) 設計インプット
ロバスト設計( タグチメソッド )による未然防止設計
要求仕様書から設計仕様書への変換
曖昧なニーズのスペックへの変換
重要な設計要素トレーサビリティーマトリックスの使用
ラベリング、梱包の設計インプット
5.820.30 ( d ) 設計アウトプット
入力のバラツキを押さえたアウトプットの確認
設計アウトプットを定義し文書類を明確にする
E-BOM / M-BOM とは
製造仕様書とは
図面発行
DMRとのトレーサビリティー
6.820.30 ( j ) 設計履歴ファイル( DHF )
DHF / DMR / DHR の定義を理解する
識別、ファイルの基本
文書体系と設計基準
7.820.30 ( e ) 設計審査
問題の早期発見、解決が目的で、セレモニーで決して終わらない
設計審査で設計のロバストネスを確認
設計審査とテクニカルレビュー
審査者がキー
8.820.30 ( f ) 設計検証
設計検証と設計バリデーションの相違
インプットからアウトプットまでトレーサブルであるとは
エビデンスに基づく設計
設計検証レポートは、設計者の活動記録
9.820.30 ( g ) 設計バリデーション
設計バリデーションの手法
社内、臨床評価の相違と記録
ソフトウェアバリデーションとVモデル
3種類のバリデーションの相違
10.820.30 ( g ) 設計移管
設計部門が製造工程を設計する
工程V&VとQC工程表
機能展開とPFMEA
製造仕様書への展開
DHF,DMR,DHR
11.820.30 ( h ) 設計変更
重要な査察ポイント
設計変更プロセスとリスクアセスメント
コミュニケーションエラー
製品不良を再発させない重要な要素、設計CAPA
文書、図面の変更管理
* 内容が変更される場合がございます。
FDA GMP( QSR )2日間コース Rev.11
〜 医療機器・IVDに求められる規制要求事項の本質を学ぶ実践コース 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
終了しました!
☆ 開催概要 ☆
グローバルに進出しようとする企業にとって 米国は最も大きなマーケットであり、米国市場での成功がグローバルを制すといっても過言ではありません。しかし、米国に上市するには医療機器規制QSRに基づくFDAの査察は避けられません。クオリス・イノーバのQSRコースは、多くのFDA査察準備、対応の経験や品質管理の経験から、FDA規制のみならず医療機器開発・製造の本質を鋭く説き、多くの企業の皆様の共感を集め、支持されて参りました。米国に進出する企業のみならず、医療機器・IVD を開発しようとしている企業の皆様のマインドが大きく変わる価値あるクオリス・イノーバのメインコースです。
日 時:2013年10月28日(月)、29日(火) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
受講料: ¥90,000( 2日割引適用、テキスト・規制邦訳文・昼食代含む )
割 引: ¥76,000( 2日分、テキスト・規制邦訳文・昼食代含む )
1社2名以上ご参加の場合
特 割: 企業で行うオンサイトセミナー開催の後、継続研修などでご利用の場合、
特別割引を適用させて頂きますのでメールでご相談下さい。
この場合、正規料金をお支払いの方が優先となり、ご希望に添えない場合が
ございますので予めご了承ください。
定 員: 30名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
第1日目 セミナー内容
A.最新情報の解説
1.FDA規制の本質( 医療機器開発・製造の本質を理解する )
2.FDA査察の本質、TQMを理解する
3.なぜISOではFDA査察にパスしないのか
4.ISOマインドを変える
5,QSITとは
6.医療機器規制を正しく理解する為に
7.QSR規制要求事項の本質を理解する
820.20 経営者の責任
経営者がリードするTQMを理解すると見えてくるマネジメントレビューの本質
FDA が一番確認したいこととは。。
820.22 品質監査
QSITに基づく効果的な監査手法と監査報告のあり方
820.25 要員
教育の重要性:教育の手法と記録、力量表、リソースとは
820.30 設計管理
ライフサイクル製品開発手法、ロバスト設計
設計検証と設計バリデーションの相違
ISO にない設計移管、工程設計とは
820.40 文書管理
グロバールに通用する文書管理手法
820.50 購買管理
これからの査察の潮流、サプライヤー管理とは
購買チームが品質に貢献するために
820.60 識別
混同を防止する為の識別方法とは
820.65 トレーサビリティー
体内埋込型機器だけではなく。。
820.80 受領、工程内及び完成品の受入
Receiving, In-process, and Finished device acceptance の相違
820.86 受入の状態
状態表示する意味
820.90 不適合品
不適合品の処理プロセス
FDAはなぜNCを見たがるのか
グローバル用語の定義を使用する
820.120 機器のラベリング
リコールが一番多く、査察で最も注目されるラベリングの管理とは
ラベルとラベリングの相違
820.130 機器の梱包
梱包箱を見ればどんな設計を行っていたかが判る
820.140 取り扱い
混同・劣化・損傷・汚染を発生させないためのリスク管理
820.150 保管
倉庫などの保管エリアのリスク管理
リスクアセスメントをすると。。
820.160 流通
劣化品の流通を防ぐ設計品質とは
820.170 据付
インスタレーション手順書のV&V
820.180 記録
重要な Objective Evidence を示すための記録管理
820.181 機器原簿(DMR)
DMRを定義し識別する
DHF,DMR,DHRの相違
820.184 機器履歴簿(DHR)
製造記録の管理方法、特に重要な。。
820.186 品質システム記録
DHF,DMR,DHR以外の品質記録とは
820.200 付帯サービス
サービス記録の分析とMR,MDRの判定
820.250 統計的手法
TQMを進めていく上で最も重要な要素
品質改善に欠かせない統計的手法
第2日目 セミナー内容
820.70 生産及び工程管理
工程を監視、管理する工程パラメータとは
工程設計と工程V&V
工程のリスクマネジメント(PFMEA)
要員、環境、汚染管理
設備の妥当性とは
保守と検査の相違
副資材の扱い
820.72 検査・測定及び試験装置
意図した用途への適合性、測定器の精度管理とは
測定器の精度管理と救済処置
820.75 工程の妥当性確認
QSR,医療機器規制の本質、工程バリデーションとは
TQMが目指すべき試験レスとは
バリデーションの手法( VMP,DQ、IQ、OQ、PQ )
設備バリデーションとは
CSV( コンピュータシステムバリデーション )とは
820.198 苦情ファイル
査察のキーポイント、苦情管理の目的を理解する
820.100 是正処置及び予防処置( CAPA )
FDA警告書で最も多い指摘CAPAの原則を理解する
修正、是正、予防の相違を明確に理解しマインドを変える
CAPA7ステップを理解する
CAPAの効果(再発防止)を出す仕組み
MRで監視すべきCAPAプロセス
( 内容が変更になる場合もございますが大きな変更はございません。)
FDA プロセスバリデーションコース Rev.8
〜 プロセスバリデーションの本質の理解と製品品質向上 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
満席につき募集を終了いたしました!
次回のご参加をお待ちしております。
☆ 開催概要 ☆
クオリス・イノーバがこれまで実施してきたGAP監査では、プロセスバリデーションのエビデンスが提出できず、手順さえもない企業がほとんどです。エビデンスを提出できたとしてもプロセスバリデーションの本質を理解していないので、監査に耐えられる満足のいくものではありません。品質システムが目指すものは、意図した用途に適合する製品を終始一貫して生産すること。これらの原則と目的を達成するための重要な要素がプロセスバリデーションです。そこで、規制当局はプロセスバリデーションの手順と結果を確認しようとします。FDA査察の結果を統計的にみてもワースト10に入る指摘項目となっています。このコースでは製品品質のバラツキを無くして品質コストを下げ、医療機器の製品安全性に貢献するプロセスバリデーションの本質を理解します。
日 時:2013年7月8日(月曜) 12:30 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している企業
FDA査察準備を考えている企業
受講料: ¥39,000
割 引: ¥29,000
1社2名以上ご参加の場合、或いは
QSRコース(4月15、16日)、設計監理コース(5月13日)を受講された方
特 割: すでに多くの方が受講されている企業様、オンサイトセミナーの後、継続研修などで
ご利用の場合、特別割引を適用させて頂きますのでご相談下さい。
正規料金をお支払い頂く方が優先され出席戴けない場合がございますので予めご了解
下さい。
定 員: 30名 満席になり次第締切ます
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
1.なぜ工程バリデーションを実施するのか?
プロセスバリデーションの目的と本質
工程管理の目的と本質
医療機器規制・規格を理解する
2.バリデーションすべき工程とは
バリデーションすべき工程の識別
検証工程とバリデーション工程の違い
設備バリデーションと工程バリデーション
バリデーション工程の例
3.バリデーションの種類
バリデーションの種類
設備バリデーションと劣化パラメータ
コンピュータシステムバリデーション(CSV)
4.バリデーションステップ
VMP: バリデーションマスタープラン
DQ: 設計時的確性評価
IQ: 据付時的確性評価
OQ: 稼働時的確性評価
PQ: 製造性能的確性評価
バリデーションの文書化
プロセスバリデーションの明確化
自動化工程の明確化
CSVカテゴリーの明確化
ソフトウェアバリデーションの基本
カテゴリー別ソフトウェア
5.設計移管とプロセスバリデーション
設計移管とプロセスバリデーションの関係
ミニスケールとフルスケール
工程設計表とトレーサビリティー
6.バリデーションの実践
マスターバリデーションプラン
バリデーション工程の識別
バリデーション計画
設備リストとバリデーション工程リスト
チームビルディング
DQ Plan /DQ Report
IQ Plan/IQ Report
OQ Plan/OQ Report
PQ Plan/PQ Report
7.バリデーションのアウトプット
QC工程表と管理図
工程能力
再バリデーションの時期設定
設備メンテナンス手順
*内容は変更される場合がございます*

CAPA コース Rev.11
〜 査察で最も指摘の多いCAPAの正しい理解と問題を再発させない仕組み 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
終了致しました!
☆ 開催概要 ☆
FDA査察の結果を統計的にまとめてみると、CAPAで最も多くの指摘をしていることが判ります。FDAは自らの査察を通して企業で最も理解されていないプロセスをよく理解して、不適合の再発原因がCAPAにあることを知っています。このことはクオリス・イノーバが実施してきたGAP監査からも明かで、日本のほとんどの企業でCAPAを正しく理解しておらずマネジメントできていません。医療機器だけではなく、医薬品や多くの業種で理解すべき内容です。FDAの査察を受けた後のアクションレターもCAPAのステップで回答すべきです。このコースに参加指されたほとんどの方のCAPAのマインドが変わる価値あるコースです。
日 時:2013年5月27日(月曜) 12:30 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVDに限らず医薬品など広くCAPAを用いた品質改善を期待される企業
受講料: ¥39,000
割 引: ¥29,000
1社2名以上ご参加の場合、或いは
QSRコース(4月15、16日)、設計監理コース(5月13日)を受講された方
特 割: すでに多くの方が受講されている企業様、オンサイトセミナーの後、継続研修などで
ご利用の場合、特別割引を適用させて頂きますのでご相談下さい。
正規料金をお支払い頂く方が優先され出席戴けない場合がございますので予めご了解
下さい。
定 員: 30名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
1.なぜ事故は再発するのか?
2.会社の文化、価値観が重要な要素
3.規制・ISO規格を理解する
4.ガイドラインの理解
5.TQMの理解と仕組みが無いとCAPAは回らない
7.再発する品質不良
8.修正・是正・予防の正しい理解
修正・CA・PAの相違
9.医療機器規制要求事項
FDA 21CFR Part 820.100 CAPA とISO 13485 8.5 改善
10.不適合、逸脱、特別採用と逸脱許可の相違
11.修正、手直し、修理の相違
12.苦情処理、不適合処理、CAPA手順との関係
13.修正プロセス
14.CA、PAエスカレーション
15.CAPA7ステップ
Six Sigma との相違
Step 1 問題を検出し明確にする
Step 2 問題の原因を詳しく調査する
Step 3 根本原因を特定する
Step 4 是正又は予防するための処置
Step 5 処置の結果を文書化
Step 6 処置の検証・妥当性確認
Step 7 有効性の確認
16.CAPAのマネジメント
17.設計CAPA
18.Warning Letter Case Study
*内容は変更される場合がございます*
昨年度講習風景

FDA設計管理ガイダンスコース Rev.10
〜 設計管理規制だけではなく会社に利益をもたらすものづくりの本質 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
満席につき募集を終了いたしました!
次回のご参加をお待ちしております。
☆ 開催概要 ☆
日本企業のほとんどの開発、設計管理の仕組みがFDAが要求する規制に準拠していません。これはグローバルに進出しようとする企業だけではなく、患者の命を預かる医療機器メーカーにとって大きなリスクを伴います。FDAは自らの査察で、医療機器品質不良の根本原因を統計的に設計管理であると見抜いており、それはクオリス・イノーバの多くのGAP監査経験でも明かです。グローバルな開発・設計基準を「FDA設計管理ガイダンス」と、欧米で取り入れられている仕組みを元に体系的に解説し、医療機器開発の本質である品質不良を発生させない仕組み(ロバスト設計)をタグチメソッドを用いて解説します。ただ規制だからではなく、設計品質を高める為に。エンジニアの方から多くの共感と支持を集めてきたクオリス・イノーバの最も人気のある価値あるコースです。
日 時:2013年5月13日(月曜) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にソフトウェア搭載機器でこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している企業
FDA査察準備を考えている企業
受講料: ¥48,000( テキスト・規制邦訳文・昼食代含む )
割 引: ¥38,000( テキスト・規制邦訳文・昼食代含む )
1社2名以上ご参加の場合、或いはQSRコース(4月15、16日)を受講された方
特 割: すでに多くの方が受講されている企業様、オンサイトセミナーの後、継続研修などで
ご利用の場合、特別割引を適用させて頂きますのでご相談下さい。
正規料金をお支払い頂く方が優先され、人気コースの為ご出席戴けない場合が
ございますので予めご了解下さい。
定 員: 40名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
1.設計品質の本質的価値
繰り返される設計品質に起因する製品不良
ISOとQSRの本質的な違い
FDAが望んでいるTQMの本質、品質工学に基づくロバスト設計
医療機器の設計手法( Water Fall Model )
一貫性を問うFDA査察
〜 TQM、FDA設計管理ガイダンスに基づくQSRサブパートCの実践 〜
2.820.30 ( a ) 総則
TQMに欠かせないマネジメントチームの重要な役割
要員教育の重要性( 820.25 )
3.820.30 ( b ) 設計及び開発計画
医療機器開発ライフサイクルマネジメント
タグチメソッドの開発方式によるフロントローディング手法
PM,PLの役割
カスタマーニーズを要求仕様に変換する品質機能展開
コンカレントエンジニアリング
4.820.30 ( c ) 設計インプット
ロバスト設計( タグチメソッド )による未然防止設計
要求仕様書から開発仕様書への変換
曖昧なニーズのスペックへの変換
重要な設計要素トレーサビリティーマトリックスの使用
5.820.30 ( d ) 設計アウトプット
入力のバラツキを押さえたアウトプットの確認
設計アウトプットを定義し文書類を明確にする
製造仕様書とは
DMRとのトレーサビリティー
6.820.30 ( e ) 設計審査
問題の早期発見、解決が目的で、セレモニーで決して終わらない
設計審査で設計のロバストネスを確認
審査者がキー
7.820.30 ( f ) 設計検証
設計検証と設計バリデーションの相違
インプットからアウトプットまでトレーサブルであるとは
設計検証レポートは、設計者の活動記録
ソフトウェアバリデーションとVモデル
8.820.30 ( g ) 設計バリデーション
設計バリデーションの手法
ソフトウェアバリデーションとVモデル
3種類のバリデーションの相違
9.820.30 ( g ) 設計移管
設計部門が製造工程を設計する
工程V&VとQC工程表
機能展開とPFMEA
製造仕様書への展開
DHF,DMR,DHR
10.820.30 ( h ) 設計変更
重要な査察ポイント
設計変更プロセスとリスクアセスメント
コミュニケーションエラー
製品不良を再発させない重要な要素、設計CAPA
* 内容が変更される場合がございます。

FDA GMP( QSR )2日間コース Rev.10
〜 医療機器・IVDに求められる規制要求事項の本質を学ぶ実践コース 〜
このコースの売り上げ金の一部は人道支援活動に寄付されます
満席につき募集を終了致しました!
次回のご参加をお待ちしております。
☆ 開催概要 ☆
グローバルに進出しようとする企業にとって 米国は最も大きなマーケットであり、米国市場での成功がグローバルを制すといっても過言ではありません。しかし、米国に上市するには医療機器規制QSRに基づくFDAの査察は避けられません。クオリス・イノーバのQSRコースは、多くのFDA査察準備、対応の経験や品質管理の経験から、FDA規制のみならず医療機器開発・製造の本質を鋭く説き、多くの企業の皆様の共感を集め、支持されて参りました。米国に進出する企業のみならず、医療機器・IVD を開発しようとしている企業の皆様のマインドが大きく変わる価値あるクオリス・イノーバのメインコースです。
日 時:2013年4月15日(月)、16日(火) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している、輸出しようとしている企業
米国FDA査察準備を考えている企業
受講料: ¥90,000( 2日割引適用、テキスト・規制邦訳文・昼食代含む )
割 引: ¥76,000( 2日分、テキスト・規制邦訳文・昼食代含む )
1社2名以上ご参加の場合
特 割: 企業で行うオンサイトセミナー開催の後、継続研修などでご利用の場合、
特別割引を適用させて頂きますのでメールでご相談下さい。
この場合、正規料金をお支払いの方が優先となり、ご希望に添えない場合が
ございますので予めご了承ください。
定 員: 30名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
第1日目 セミナー内容
A.最新情報の解説
1.FDA規制の本質( 医療機器開発・製造の本質を理解する )
2.FDA査察の本質、TQMを理解する
3.なぜISOではFDA査察にパスしないのか
4.ISOマインドを変える
5,QSITとは
6.医療機器規制を正しく理解する為に
7.QSR規制要求事項の本質を理解する
820.20 経営者の責任
経営者がリードするTQMを理解すると見えてくるマネジメントレビューの本質
FDA が一番確認したいこととは。。
820.22 品質監査
QSITに基づく効果的な監査手法と監査報告のあり方
820.25 要員
教育の重要性:教育の手法と記録、力量表とは
820.30 設計管理
ライフサイクル製品開発手法、ロバスト設計
設計検証と設計バリデーションの相違
設計移管、工程設計とは
820.40 文書管理
グロバールに通用する文書管理手法
820.50 購買管理
これからの査察の潮流、サプライヤー管理とは
購買チームが品質に貢献するために
820.60 識別
混同を防止する為の識別方法とは
820.65 トレーサビリティー
体内埋込型機器だけではなく。。
820.80 受領、工程内及び完成品の受入
Receiving, In-process, and Finished device acceptance の違いは
820.86 受入の状態
状態表示する意味
820.90 不適合品
不適合品の処理プロセス
FDAはなぜNCを見たがるのか
グローバル用語の定義を使用する
820.120 機器のラベリング
リコールが一番多く、査察で最も注目されるラベリングの管理とは
ラベルとラベリングの相違
820.130 機器の梱包
梱包箱を見ればどんな設計を行っていたかが判る
820.140 取り扱い
混同・劣化・損傷・汚染を発生させないためのリスク管理
820.150 保管
倉庫などの保管エリアのリスク管理
リスクアセスメントをすると。。
820.160 流通
劣化品の流通を防ぐ設計品質とは
820.170 据付
インスタレーション手順書のV&V
820.180 記録
重要な Objective Evidence を示すための記録管理
820.181 機器原簿(DMR)
DMRを定義し識別する
DHF,DMR,DHRの相違
820.184 機器履歴簿(DHR)
製造記録の管理方法、特に重要な。。
820.186 品質システム記録
DHF,DMR,DHR以外の品質記録とは
820.200 付帯サービス
サービス記録の分析とMR,MDRの判定
820.250 統計的手法
TQMを進めていく上で最も重要な要素
品質改善に欠かせない統計的手法
第2日目 セミナー内容
820.70 生産及び工程管理
工程を監視、管理するとは
工程設計と工程V&V
工程のリスクマネジメント(PFMEA)
820.72 検査・測定及び試験装置
意図した用途への適合性、測定器の精度管理とは
Maintenance と Inspection の相違
820.75 工程の妥当性確認
QSR,医療機器規制の本質、工程バリデーションとは
TQMが目指すべき試験レスとは
バリデーションの手法( VMP,DQ、IQ、OQ、PQ )
設備バリデーションとは
CSV( コンピュータシステムバリデーション )とは
820.198 苦情ファイル
査察のキーポイント、苦情管理の目的を理解する
820.100 是正処置及び予防処置( CAPA )
FDA警告書で最も多い指摘CAPAの原則を理解する
修正、是正、予防の相違を明確に理解しマインドを変える
CAPA7ステップを理解する
CAPAの効果(再発防止)を出す仕組み
MRで監視すべきCAPAプロセス
( 内容が変更になる場合もございますが大きな変更はございません。)

2012年度オープンセミナーのご案内
2011年度オープンセミナーには多くの方にご出席頂き誠にありがとうございました。また、2011年度のオンサイトセミナーは過去最も多く開催させて頂きました。( オンサイトセミナー( 企業内トレーニング )のお問い合わせはこちらまで ) 現在2012年度に予定されておりますセミナーの題目と内容を以下の通りご案内致します。( この予定は随時アップデートされます )開催日程などは約開催2ヶ月前にこのページでお知らせする予定です。各コースの詳細はこちらを御覧下さい。
11月開催予定でしたプロセスバリデーションコースは来年に延期となり、12月までに開催される弊社主催セミナーは今のところございません。 弊社代表による日経等のセミナーハウス主催セミナーを個別に掲載紹介致します。 今年末までに来年のセミナースケジュールを掲載予定ですが、年内までの開催をお待ち頂いた皆様方やお問い合わせを戴いた皆様に心よりお詫びを申し上げます。
( 2012_09_30 up )
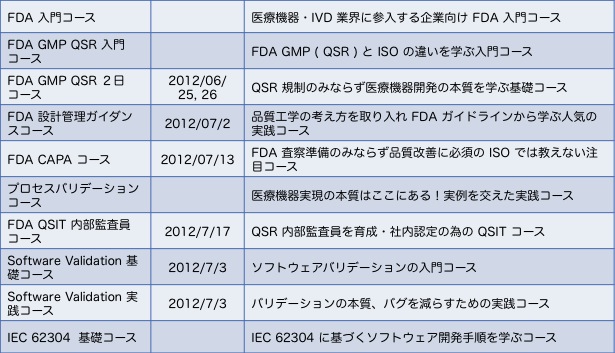

品質システムが求める文書の本質
〜 なぜ手順に従わないのか 〜
このコースは【 情報機構 】主催で開催されます。
http://www.johokiko.co.jp/seminar_medical/AA121202.php
終了致しました!
開催概要
日 時:2012年12月7日(金) 12:30 〜 16:30
会 場:タワーホール船堀3F303会議室( 東京・船堀 )
主 催:情報機構
講 師;クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に参入しているものの医療機器を想定した開発体制が未整備な企業
品質に関する査察の厳しい地域(特に米国)向けの医療機器を開発している企業、
またはこれから開発しようとしている企業
申 込:直接主催者HPからお申し込み下さい。
セミナー内容
1. 監査から見えてくるISO認証会社の実態
・手順から読み取れる
・教育プランから読み取れる力量と教育プロセス
・教育プロセスのリソースとは
・製造手順からわかる設計移管プロセスの欠如
2. 手順を見ればその会社の品質がわかる
・何を誰に伝えたいのか?
・ユーザー不在の自己中文書
・文書の検証とは
・プロセスのフレームを見せることができるか
3. FDA の文書に関わる Warning Letter ( 警告書 )
・FDA Warning Letter から理解できること
・規制が求める手順とは
・手順不備に伴うリスク
4. なぜ手順に従わないのか
・CAPAを実施する
・手順を読むカスタマーは誰か
・手順の変更に伴う教育
・力量の要件である手順
5. 手順を守らせ品質を向上させる為の手法
・ISOマインドからの脱却
・現状を理解させる
・リスクをマネジメントチームに理解させる
・教育の方法を変える
・プロセスをマネジメントレビューで評価する
医療機器新規参入の手引き
〜 製品開発の勘所を押さえる 〜
このコースは【 日経ものづくり 】主催で開催されます。
http://techon.nikkeibp.co.jp/article/SEMINAR/20120922/241351/


終了致しました!
開催概要
日 時:2012年10月29日(月) 13:30 〜 17:30
会 場:秋葉原 UDX カンファレンス( 東京・千代田区秋葉原 )
主 催:日経ものづくり
講 師;クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に参入しているものの医療機器を想定した開発体制が未整備な企業
品質に関する査察の厳しい地域(特に米国)向けの医療機器を開発している企業、
またはこれから開発しようとしている企業
申 込:直接主催者HPからお申し込み下さい。
セミナー内容
1.製品企画
2.製品開発プロセス
3.品質、製品不具合
4.規制、国際標準
5.プロジェクト推進

ソフトウェアバリデーションコース
〜 医療機器ソフトウェア開発に必須の基礎知識とその本質を理解する 〜
募集を終了致しました!
次回のご出席をお待ちしております。
☆ 開催概要 ☆
米国FDAのみならず、EU医療機器指令、中国SFDA、医療機器ソフトウェア申請基本要求等、各国の医療機器ソフトウェアに関する規制が強化されています。FDA医療機器ソフトウェアガイドライン、最新の中国SFDガイドライン、FDAモバイルアプリケーションガイドを取り入れ、医療機器規制が求める本質を理解し、開発手法を学びます。特に中国SFDAの翻訳資料だけでも価値あるコースです。
日 時: 2012年7月3日(火曜) 10:00 〜 16:30
会 場: 東京都立産業貿易センター浜松町館( 東京・浜松町 )
( JR浜松町駅から徒歩5分 )
講 師; クオリス・イノーバ株式会社
株式会社イマテック
対 象: 医療機器・IVD市場にソフトウェア搭載機器でこれから参入しようと考えている企業
既に医療機器・IVDに限らず製薬会社でも製品を海外に輸出している企業
FDA査察準備を考えている企業
FDA査察を終えた企業で指摘を受けた企業
医療機器に限らず品質問題の撲滅為の方法を模索している企業
受講料: ¥53,000( テキスト・規制、ガイドライン翻訳含む )
割 引: ¥43,000( テキスト・規制、ガイドライン翻訳含む )
1社2名以上ご参加の場合、或いはQSRコース(6月25,26日)を受講された方
ソフトウェアバリデーションコース(7月3日)を受講予定の方など7月中に開催される
コースを受講される方。
特 割: オンサイトセミナーの後、継続研修などでご利用の場合、特別割引を適用させて頂きます
のでご相談下さい。 この場合、正規料金をお支払い頂く方が優先され、ご出席戴けない
場合がございますので予めご了承下さい。
定 員: 40名
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
また複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ:
お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
1.FDAによるソフトウェアバリデーション要求
a.FDAによる医療機器ソフトウェアの規制( 邦訳付き )
b.FDAが公開するソフトウェアガイダンス( 邦訳付き )
2.ソフトウェア・バリデーションの基本
a.医療機器とソフトウェア開発ライフサイクル
b.ソフトウェア開発計画
c.ソフトウェア要求事項分析と設計
d.ソフトウェア検証(直交表を用いた組み合わせ試験等)
e.ソフトウェア妥当性確認
f.FDA Warning Letter 事例紹介(1)
3.規格・規制要求の理解
a.医療機器ソフトウェアに対する規制(US,EU,中国等)
b.IEC62304の規制( 概要邦訳付き )
c.コンピュータシステムバリデーション(CSV)
4.ソフトウェア・バリデーションの現実
a.FDA Warning Letter 事例紹介(2)
b.ソフトウェアリスクマネジメント
c.ソフトウェア構成管理
d.ソフトウェア問題解決
5.ソフトウェアに関する新しい規制
a.MDDS(医療機器データシステム)
b.Mobile Medical Application ガイドライン
c. IEC 80001-1 医療機器を組込むITネットワークのためのリスクマネジメントの適用
Appendix( 別冊翻訳本 )
A1:ソフトウェアバリデーションの一般原則( 製造業者及びFDAスタッフのためのガイダンス )
A2:医療機器に含まれるソフトウェアのための市販前申請の内容に関するガイダンス
A3:医療機器に於ける規制ソフトウェアの使用に関する企業、FDA査察官及び適合性の為の指針
A4:IEC 62304 の規格概要
A5:医薬品・医療部外品製造販売業者等におけるコンピュータ化システム適正管理ガイドライン
( 厚生労働省通知 )
A6 : 中国SFDA 医療機器ソフトウェア申請基本要求ガイドライン
FDA CAPA Rev.10 コース
〜 査察で最も指摘の多いCAPAの正しい理解と問題を再発させない仕組み 〜
終了いたしました!
☆ 開催概要 ☆
FDA査察の結果を統計的にまとめてみると、CAPAで最も多くの指摘をしていることが判ります。FDAは自らの査察を通して企業で最も理解されていないプロセスをよく理解して、品質不良の再発の原因がCAPAにあることを知っています。このことはクオリス・イノーバが実施してきたGAP監査からも明かで、日本のほとんどの企業でCAPAを正しく理解しておらず、マネジメントできていません。FDAの査察を受けた後のアクションレターもCAPAのステップで回答すべきです。このコースに参加指されたほとんどの方のCAPAに対する考え方が変わりマインドが変わる価値あるコースです。
日 時:2012年7月13日(金曜) 10:00 〜 16:00
会 場:東京都立産業貿易センター浜松町館( 東京・浜松町 )
( JR浜松町駅から徒歩5分 )
対 象:医療機器・IVD市場にソフトウェア搭載機器でこれから参入しようと考えている企業
既に医療機器・IVDに限らず製薬会社でも製品を海外に輸出している企業
FDA査察準備を考えている企業
FDA査察を終えた企業で指摘を受けた企業
医療機器に限らず品質問題の撲滅為の方法を模索している企業
受講料: ¥48,000( テキスト・昼食代含む )
割 引: ¥38,000( テキスト・昼食代含む )
1社2名以上ご参加の場合、或いはQSRコース(6月25,26日)を受講された方
ソフトウェアバリデーションコース(7月3日)を受講予定の方など7月中に開催される
コースを受講される方。
特 割: すでに多くの方が受講されている企業様、オンサイトセミナーの後、継続研修などで
ご利用の場合、特別割引を適用させて頂きますのでご相談下さい。
正規料金をお支払い頂く方が優先されご出席戴けない場合がございますので予め
ご了承下さい。
定 員: 30名
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
6、7月に弊社コースを受講される方は日付とコース名を問い合わせ欄にご記入下さい。
また複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
1.なぜ事故は再発するのか?
2.会社の文化、価値観が重要な要素
3.医療機器規制を理解する
4.ガイドラインの理解
5.TQMの仕組みが無いとCAPAは回らない
6.グローバル規格・ガイダンスを理解する
7.再発する品質不良
8.修正・是正・予防の正しい理解
CA・PAの相違
修正・CAの相違
修正・PAの相違
9.規制要求事項 Part 820.100
10.CA/PAエスカレーション
11.CAエスカレーション
12.PAエスカレーション
13.CAPA7ステップ
Six Sigma との相違
Step 1 問題を検出し明確にする
Step 2 問題の原因を詳しく調査する
Step 3 根本原因を特定する
Step 4 是正又は予防するための処置
Step 5 処置の結果を文書化
Step 6 処置の検証・妥当性確認
Step 7 有効性の確認
14.CAPAのマネジメント
15.不適合・CAPAフロー
16.設計CAPA
17.Warning Letter Case Study
QSIT 監査手法コース
〜 QSR内部監査員養成の為の社内資格認定コース 〜
終了致しました!
☆ 開催概要 ☆
FDA QSRの内部監査員を養成するコースです。お客様からご要望の多かった社内監査員に力量を与える要件となるコースとなるでしょう。品質システムがうまく回っていないのは、内部監査員がその本質をきちんと見抜いていなからです。しかし、片手間に行っている社内内部監査員のレベルを上げるのは並大抵なことではありません。このコースではグローバル監査を経験しFDA査察をよく知るクオリス・イノーバならではの監査の目の付け所(ノウハウ)を品質管理の本質を元に解説する実践コースです。
コース終了後、試験を実施し基準を満たした方に修了書をお送りします。
日 時:2012年7月17日(火曜) 10:00 〜 16:30
会 場:東京都立産業貿易センター浜松町館( 東京・浜松町 )
( JR浜松町駅から徒歩5分 )
対 象:すでに当社QSRコースをオープン、オンサイトを問わず受講されている方限定です。
( QSRの本質を理解していないで監査員は務まらず、適切な監査は期待できません。
本コースはQSRを理解している事を前提としております。まだ受講されていない方は
6月25,26日開催のQSRコースを受講願います。 FDA入門コースでは受講で
きません。 )
受講料: ¥48,000( テキスト・ガイド邦訳文・昼食代含む )
割 引: ¥38,000( テキスト・ガイド邦訳文・昼食代含む )
1社2名以上ご参加の場合、或いはQSRコース(6月25,26日)を受講された方
ソフトウェアバリデーションコース(7月3日)を受講予定の方など7月中に開催される
コースを受講される方。
定 員: 30名
( 修了試験の基準を満たした方に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
過去に受講された弊社QSRコースの受講日を問い合わせ欄に記載して下さい。
オンサイトセミナーでも構いません。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
1.監査を計画する
2.CAPAの監査は全部門で実施する
3.手順を説明させて記録を確認する( 事実の把握 )
4.品質に貢献する管理パラメータは何か
5.各部門・機能の役割を理解する
6.各パート 監査の目のつけどころ
1.経営者による管理
2.設計管理
3.是正及び予防処置(CAPA)
MDR報告
改修及び回収(CAR)
医療機器トラッキング(MDT)
4.生産及び工程管理(P&PC)
5.滅菌工程(SPC)
FDA設計管理ガイダンスコース Rev.9
〜 設計管理規制だけではなく会社にとって利益をもたらすものづくりの本質 〜
満席につき募集を終了致しました!
次回のご出席をお待ちしております。
☆ 開催概要 ☆
日本のほとんどの企業において、開発、設計管理の仕組みがFDAが要求する規制に準拠していません。これはグローバルに進出しようとする企業にとって大きなリスクを伴います。FDAは自らの査察で、医療機器品質不良の根本原因を統計的に設計管理であると見抜いており、それはクオリス・イノーバのGAP監査でもあきらかです。グローバルな開発・設計基準を「FDA設計管理ガイダンス」と欧米で取り入れられている仕組みを元に体系的に解説し、医療機器開発の本質である品質不良を発生させない仕組み(ロバスト設計)をタグチメソッドを用いて解説します。エンジニアの方から多くの共感と支持を集めてきたクオリス・イノーバの最も人気のある価値あるコースです。
日 時:2012年7月2日(月曜) 10:00 〜 16:30
会 場:東京都立産業貿易センター浜松町館( 東京・浜松町 )
( JR浜松町駅から徒歩5分 )
対 象:医療機器・IVD市場にソフトウェア搭載機器でこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している企業
FDA査察準備を考えている企業
受講料: ¥48,000( テキスト・規制邦訳文・昼食代含む )
割 引: ¥38,000( テキスト・規制邦訳文・昼食代含む )
1社2名以上ご参加の場合、或いはQSRコース(6月25,26日)を受講された方
ソフトウェアバリデーションコース(7月3日)を受講予定の方など7月中に開催される
コースを受講される方。
特 割: すでに多くの方が受講されている企業様、オンサイトセミナーの後、継続研修などで
ご利用の場合、特別割引を適用させて頂きますのでご相談下さい。
正規料金をお支払い頂く方が優先され、人気コースの為ご出席戴けない場合が
ございますので予めご了解下さい。
聴 講: 過去本コースを受講された方が対象です。
再度受講されたい方は格安な聴講料金でご参加できます。
正規料金をお支払い頂く方が優先され、人気コースの為ご出席戴けない場合が
ございますので予めご了解下さい。
定 員: 40名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
1.設計品質の本質的価値
繰り返される設計品質に起因する製品不良
ISOとQSRの本質的な違い
FDAが望んでいるTQMの本質、品質工学に基づくロバスト設計
医療機器の設計手法( Water Fall Model )
一貫性を問うFDA査察
〜 TQM、FDA設計管理ガイダンスに基づくQSRサブパートCの実践 〜
2.820.30 ( a ) 総則
TQMに欠かせないマネジメントチームの重要な役割
要員教育の重要性( 820.25 )
3.820.30 ( b ) 設計及び開発計画
医療機器開発ライフサイクルマネジメント
タグチメソッドの開発方式によるフロントローディング手法
PM,PLの役割
カスタマーニーズを要求仕様に変換する品質機能展開
コンカレントエンジニアリング
4.820.30 ( c ) 設計インプット
ロバスト設計( タグチメソッド )による未然防止設計
要求仕様書から開発仕様書への変換
曖昧なニーズのスペックへの変換
重要な設計要素トレーサビリティーマトリックスの使用
5.820.30 ( d ) 設計アウトプット
入力のバラツキを押さえたアウトプットの確認
設計アウトプットを定義し文書類を明確にする
製造仕様書とは
DMRとのトレーサビリティー
6.820.30 ( e ) 設計審査
問題の早期発見、解決が目的で、セレモニーで決して終わらない
設計審査で設計のロバストネスを確認
審査者がキー
7.820.30 ( f ) 設計検証
設計検証と設計バリデーションの相違
インプットからアウトプットまでトレーサブルであるとは
設計検証レポートは、設計者の活動記録
ソフトウェアバリデーションとVモデル
8.820.30 ( g ) 設計バリデーション
設計バリデーションの手法
ソフトウェアバリデーションとVモデル
3種類のバリデーションの相違
9.820.30 ( g ) 設計移管
設計部門が製造工程を設計する
工程V&VとQC工程表
機能展開とPFMEA
製造仕様書への展開
DHF,DMR,DHR
10.820.30 ( h ) 設計変更
重要な査察ポイント
設計変更プロセスとリスクアセスメント
コミュニケーションエラー
製品不良を再発させない重要な要素、設計CAPA
FDA GMP( QSR )2日間コース Rev.9
〜 医療機器・IVDに求められる規制要求事項の本質を学ぶ実践コース 〜
満席につき募集を終了致しました!
次回のご出席をお待ちしております。
☆ 開催概要 ☆
グローバルに進出しようとする企業にとって 米国は最も大きなマーケットであり、米国の医療機器規制QSRに基づくFDAの査察は避けられません。クオリス・イノーバのQSRコースは、多くのFDA査察準備、対応の経験や、品質管理の経験から、FDA規制のみならず、医療機器開発・製造の本質を鋭く説き、多くの企業の皆様の共感を集め、支持されて参りました。出席者のマインドが大きく変わる価値あるクオリス・イノーバのメインコースです。
日 時:2012年6月25日(月)、26日(火) 10:00 〜 16:30
会 場:全国町村会館( 東京・永田町 )
( 永田町から徒歩1分 )
対 象:医療機器・IVD市場にソフトウェア搭載機器でこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している企業
FDA査察準備を考えている企業
受講料: ¥90,000( 2日分、テキスト・規制邦訳文・昼食代含む )
割 引: ¥79,000( 2日分、テキスト・規制邦訳文・昼食代含む )
1社2名以上ご参加の場合、或いは続けて設計管理コース(7月2日)を受講される方
特 割: すでに多くの方が受講されている企業様、オンサイトセミナーの後、継続研修などで
ご利用の場合、特別割引を適用させて頂きますのでご相談下さい。
満席の場合は正規料金をお支払いの方が優先となりますので予めご了承ください。
定 員: 30名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
☆ セミナー内容 ☆
第1日目 セミナー内容
1.FDA規制の本質( 医療機器開発・製造の本質を理解する )
2.FDA査察の本質、TQMを理解する
3.なぜISOではFDA査察にパスしないのか
4.ISOマインドを変える
5,QSITとは
6.医療機器規制を正しく理解する為に
7.QSR規制要求事項の本質を理解する
820.20 経営者の責任
経営者がリードするTQMを理解すると見えてくるMRの本質
820.22 品質監査
QSITに基づく効果的な監査手法と監査報告のあり方
820.25 要員
教育の重要性:教育の手法と記録
820.30 設計管理
ライフサイクル製品開発手法、ロバスト設計
設計検証と設計バリデーションの相違
820.40 文書管理
グロバールに通用する文書管理
820.50 購買管理
これからの査察の潮流、サプライヤー管理とは
購買チームが品質に貢献するために
820.60 識別
混同を防止する為の識別方法とは
820.65 トレーサビリティー
体内埋込型機器だけではなく。。
820.80 受領、工程内及び完成品の受入
受入試験方法、完成品の管理方法
820.86 受入の状態
状態表示する意味
820.90 不適合品
不適合品の処理と品質管理
FDAはなぜNCを見たがるのか
820.120 機器のラベリング
リコールが一番多く、査察で最も注目されるラベリングとは
820.130 機器の梱包
梱包箱を見ればどんな設計を行っていたかが判る
820.140 取り扱い
混同・劣化・損傷・汚染を発生させないためのリスク管理
820.150 保管
倉庫などの保管エリアのリスク管理
820.160 流通
劣化品の流通を防ぐ品質管理とは
820.170 据付
インスタレーション手順書のV&V
820.180 記録
Objective Evidence を示すための記録管理
820.181 機器原簿(DMR)
DMRを定義し識別する
820.184 機器履歴簿(DHR)
製造記録の管理方法
820.186 品質システム記録
DHF,DHR以外の品質記録とは
820.200 付帯サービス
サービス記録の分析とMR,MDR
820.250 統計的手法
TQMを進めていく上で最も重要な要素
第2日目 セミナー内容
820.70 生産及び工程管理
工程を監視、管理するとは
工程設計と工程V&V
工程のリスクマネジメント(PFMEA)
820.72 検査・測定及び試験装置
意図した用途への適合性、測定器の精度管理とは
820.75 工程の妥当性確認
QSR,医療機器規制の本質、工程バリデーションとは
TQMが目指すべき試験レスとは
バリデーションの手法( VMP,DQ、IQ、OQ、PQ )
設備バリデーションとは
CSV( コンピュータシステムバリデーション )とは
820.198 苦情ファイル
査察のキーポイント、苦情の管理の目的を理解する
820.100 是正処置及び予防処置
ウォーニングレターで最も多い指摘CAPAの原則を理解する
修正、是正、予防の相違を明確に理解しマインドを変える
CAPA7ステップを理解する
CAPAの効果的なマネジメントと予防

FDA QSR 入門コース
〜 なぜISOではFDA査察に通用しないのか 〜
このコースは【 情報機構 】主催で開催されます。
http://www.johokiko.co.jp/seminar_medical/AA120401.php
開催概要
日 時:2012年4月13日(金) 12:30 〜 16:30 終了しました!
会 場:きゅりあん4F第2特別講習室( 東京・大井町 )
主 催:情報機構
講 師;クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象:医療機器・IVD市場にすでに参入している企業で米国へ輸出を検討している方
申 込:直接主催者HPからお申し込み下さい。
セミナー内容:
1.概要
医療機器・IVD(体外診断薬)は、国内市場だけでビジネスを行う時代ではなく、グローバルでビジネスを行わなければならず、特に世界で最も市場の大きな米国の市場は無視できません。しかし、このドメインでは各国規制の枠組みの中でしかビジネスを行う事ができず、患者の命に直結する製品の品質には、特に厳しい品質システムに関わる規制が施行されています。
グローバルでは、ISO13485(ISO9001のセクター規格)に準拠することを求められますが、米国でISOではなく、QSR(品質医システム規制)に準拠することを求められます。米国で医療機器を所轄するFDA(米国食品医薬品局)は、QSRはISOから作られたものであると公言していますが、最も厳しいと言われるFDAの査察にISOを取得している企業に次々と警告書が出され、出荷停止に追い込まれている企業や、最悪のケースで何十億もの罰金を科せられた企業さえあります。その裏にはISOすら満足に準拠していない実態が見えてきます。
2.内容
1. Compliance / Integrity
2. 人の命を守る医療機器
3. 繰り返す事故・不具合
4. ISO企業の実態
5. マネジメントチーム主導のTQM
6. 行政によるミッションの相違
7. 医療機器規制の理解
8. 用語を正しく理解する
9. QSRを理解する為のガイドライン
10. TQMの最重要要素CAPA
11. 内部監査はエキスパートが行う
12. 教育の重要性
13. 文書管理の重要性
14. 初期設計で決まる製品品質
15. 製品開発は一貫性を示す
16. 設計V&Vの理解
17. 記録の重要性( DHF/DMR/DHR )
18. 設計移管は設計の責任範囲
19. 工程バリデーション
20. CSVの適用
21. ラベリングの重要性
22. 梱包は設計管理の責任
23. 統計的手法とTQM
医療機器の品質問題を防ぐ仕組みと開発手法
〜 ISOの正しい解釈を医療機器規制と事故事例に学ぶ 〜
このコースは【 日経ものづくり 】主催で開催されます。
http://techon.nikkeibp.co.jp/article/SEMINAR/20120206/204431/
開催概要
日 時:2012年3月26日(月) 10:00 〜 17:00 終了しました!
会 場:Learning Square 新橋( 東京・新橋 )
主 催:日経ものづくり
講 師;クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象:医療機器・IVD市場にすでに参入している企業、
これから参入を予定している企業の設計・生産技術、品質に関わる方
申 込:直接主催者HPからお申し込み下さい。
セミナー内容:
1.なぜ医療機器の品質問題がなくならないのか
「あってはならない」とされる医療機器の品質問題だが、実際にはなかなか減っていない。食品医薬品局(FDA)の査察が厳しい米国では、名だたる大メーカーがリコールに追われている。メーカーも手を打っているはずなのに、なぜ医療機器の品質問題がなくならないのか、一般的な機器の事故事例を基に解説する。
2.品質問題の直接原因と根本原因を区別する
品質問題がなかなか減らない理由の1つに、根本原因までさかのぼって分析していないことが挙げられる。多くの場合、直接原因の特定だけで満足してしまっているため、同じような品質問題を繰り返すことになるのだ。直接原因と根本原因の違いを明らかにした上で、根本原因を見つけるための考え方を紹介する。
3.CAPAとは何か
品質問題を根絶するには、開発体制の問題点を完全に取り除いて品質問題の発生を未然に防ぐCAPA( Corrective Action & Preventive Action、是正処置及び予防処置 )の取組みが欠かせない。しかし、多くの企業では単なる直接原因の"モグラ叩き”に終始している。CAPAの真髄を解説する。
4.製品開発にCAPAを活かす
CAPAのアプローチを自社の製品開発に導入するには、どうすればよいのか。当然、自社のやり方を見直さなければならない部分が出てくる。医療機器メーカー、部品・材料メーカーなどそれぞれの立場に向けて、CAPAを導入する上で基本となる考え方を紹介する。
5.品質問題の事例に学ぶ
直接原因と根本原因の違いを正確に区別し、根本原因までだかのぼって分析できるようになるには、過去の品質問題に学ぶ事が有効である。医療機器に限らずさまざまな事例を題材として取り上げることで、本質を見分けるための"目”を養う。ここでは、『日経ものづくり』のコラム「事故は語る」や、同コラムをまとめた書籍『事故の事典』も活用する。

医療機器 ソフトウェアバリデーションコース
〜 FDA ガイドラインに適合するソフトウェアバリデーションの基礎 〜
開催概要
日 時:2011年10月31日(月) 10:00 〜 16:00 終了しました!
会 場:東京都立産業貿易センター浜松町館( 東京・浜松町 )
( JR浜松町駅から徒歩5分 )
主 催:クオリス・イノーバ株式会社
講 師;今関 剛 氏( 株式会社イマテック 代表取締役・SESSAME会員 )
酒井 郁子氏( 株式会社イマテック AFFORDD運営委員・SESSAME会員 )
対 象:医療機器・IVD市場にソフトウェア搭載機器でこれから参入しようと考えている企業
医療機器ソフトウェアを受託開発しようと考えている企業
既に医療機器ソフトウェアを受託開発している企業
既に参入しているが医療機器ソフトウェアの開発体制が未整備な企業
医療機器に限らずソフトウェア品質問題撲滅のための方法を模索している企業
ISOではソフトウェアの品質問題が解決しないと嘆いている企業
受講料: ¥48,000( テキスト・規制邦訳文・昼食代含む )
割 引: ¥38,000( テキスト・規制邦訳文・昼食代含む )
1社2名以上ご参加の場合
定 員: 50名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込:セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
セミナー内容:
1.規格・規制要求の理解
a.医療機器ソフトウェアに対する規制
b.FDAによる医療機器ソフトウェアの規制( 邦訳付き )
c.FDAが公開するソフトウェアガイダンス( 邦訳付き )
d.IEC62304の規制( 概要邦訳付き )
e.コンピュータシステムバリデーション(CSV)
2.ソフトウェア・バリデーションの基本
a.医療機器とソフトウェア開発ライフサイクル
b.ソフトウェア開発計画
c.ソフトウェア要求事項分析と設計
d.ソフトウェア検証( 直交表を用いた組合せ試験など )
e.ソフトウェア妥当性確認
3.ソフトウェア・バリデーションの基本
a.FDA Warninig Letter 事例紹介
b.ソフトウェアリスクマネジメント
c.ソフトウェア構成管理
d.ソフトウェア問題解決
4.ソフトウェアに関するあたらしいFDAの規制
a.MDDS(医療機器データシステム)
b.Mobile Medical Application ガイドライン
本セミナーの配付資料にはテキストの他に下記のFDAガイダンス邦訳が添付されます。
Appendix
A1:ソフトウェアバリデーションの一般原則(製造業者及びFDAスタッフのためのガイダンス)
A2:医療機器に含まれるソフトウェアのための市販前申請の内容に関するガイダンス
A3:医療機器に於ける規制ソフトウェアの使用に関する企業、FDA査察官及び適合性の為の指針
A4:IEC 62304 の概要
A5:医薬品・医療部外品製造販売業者等におけるコンピュータ化システム適正管理ガイドライン
* A4 は規格概要、A5 は厚生労働省通知
FDA CAPA( 是正及び予防処置 )
〜 査察で最も指摘の多いCAPAの正しい理解と問題を再発させない仕組み 〜
このコースは【 情報機構 】主催で開催されます。
http://www.johokiko.co.jp/seminar_medical/AA111052.php
終了しました!
開催概要
日 時:2011年10月21日(金) 12:30 〜 16:30
会 場:ちよだ PLATFORM SQUARE ( 東京・竹橋 )
主 催:情報機構
講 師;クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に参入しているが医療機器を想定した開発体制が未整備な企業
米国に医療機器を輸出していてFDA査察準備をしている企業
医療機器に限らず品質問題撲滅のための方法を模索している企業
ISOでは品質問題が解決しないと嘆いている企業
FDA 査察で Form 483 で指摘をされレスポンスをしなければならない企業
申 込:直接主催者HPからお申し込み下さい。 こちらから。
講師割引:以前弊社オープンセミナー、オンサイトセミナーに御参加頂いた企業様は割引がございます。
お申し込みは開催日とコース名講師割引希望とご記入の上こちらからお申し込み下さい。
セミナー内容:
1.導入
a. 患者の命と会社を守る為に
b. Quality Cost 品質をお金で表現
c. 行政によるミッションの相違
d. 米国と日本の行政の相違
2.医療機器規制を理解する
a. QSRとISOの相違
b. ガイダンスの理解
c. QSIT を理解する
d. Warning Letter から理解できること
e. 日本の企業が受けた Warning Letter から理解できること
f. 要求規制とガイダンス
g. 規制を正しく理解するために
h. 規制・規格の用語を国際用語に揃える
3.再発する品質不良
a. なぜ品質不良は再発するのか?
b. FDA Expert の分析
4.用語の正しい理解
a. 3つの用語を理解する
b. 修正、是正処置、予防処置
c. 修正と是正の違い
d. 是正と予防の違い
5.規制要求事項
a. 21CFR820.100
b. 現存 or 潜在する問題の抽出
c. 再発問題の抽出
d. Warning Letter Case Study
6.CAPA7ステップ
Step 1. 問題を検出し明確にする
Step 2. 問題を詳しく調査する
Step 3. 根本原因を特定する
Step 4. 是正又は予防するための処置
Step 5. 処置の結果を文書化
Step 6. 処置の検証 or 妥当性確認を実施
Step 7. 有効性の確認
7.日常の対処方法
a. 簡易CAPAを使う
b. ディシジョンツリー
c. 潜在的問題の抽出と予防処置
8.CAPA のマネジメント
a. 品質問題は管理する
b. Warning Letter Case Study
9.事例研究
FDA GMP( QSR )2日間コース Rev.8
〜 医療機器・IVDに求められる規制要求事項の本質を学ぶ実践コース 〜
開催概要
終了致しました!
ご出席ありがとうございました。
日 時:2011年11月24日(木)、25日(金) 10:00 〜 16:30
会 場:東京都立産業貿易センター浜松町館( 東京・浜松町 )
( JR浜松町駅から徒歩5分 )
対 象:医療機器・IVD市場にソフトウェア搭載機器でこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している企業
FDA査察準備を考えている企業
受講料: ¥90,000( 2日分、テキスト・規制邦訳文・昼食代含む )
割 引: ¥79,000( 2日分、テキスト・規制邦訳文・昼食代含む )
1社2名以上ご参加の場合、続けて設計管理コース(11月30日)を受講される方
特 割: すでに多くの方が受講されている企業様、オンサイトセミナーの後、継続研修などで
ご利用の場合、特別割引を適用させて頂きますのでご相談下さい。
定 員: 30名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208

セミナー内容
第1日目 セミナー内容
1.FDA規制の本質
2.FDA査察の本質、TQMを理解する
3,QSITとは
4.規制要求事項の実践
820.20 経営者の責任
TQMを理解すると見えてくるMRの本質
820.22 品質監査
QSITに基づく監査手法と監査報告のあり方
820.25 要員
教育の重要性:教育の手法と記録
820.30 設計計画
ライフサイクル製品開発手法、ロバスト設計
設計検証と設計バリデーションの相違
820.40 文書管理
グロバールに通用する文書管理
820.50 購買管理
これからの査察の潮流、サプライヤー管理とは
820.60 識別
混同を防止する為の識別方法とは
820.65 トレーサビリティー
体内埋込型機器だけではなく。。
820.80 受領、工程内及び完成品の受入
受入試験方法、完成品の管理方法
820.86 受入の状態
状態表示する意味
820.90 不適合品
不適合品の処理と品質管理
820.120 機器のラベリング
リコールが一番多く、査察で最も注目されるラベリングとは
820.130 機器の梱包
梱包箱を見ればどんな設計を行っていたが判る
820.140 取り扱い
混同・劣化・損傷・汚染を発生させないためのリスク管理
820.150 保管
倉庫などの保管アリアのリスク管理
820.160 流通
劣化品の流通を防ぐ品質管理とは
820.170 据付
インスタレーション手順書のV&V
820.180 記録
Objective Evidence を示すための記録管理
820.181 機器原簿(DMR)
DMRを定義し識別する
820.184 機器履歴簿(DHR)
製造記録の管理方法
820.186 品質システム記録
DHF,DHR以外の品質記録とは
820.200 付帯サービス
サービス記録の分析とMR,MDR
820.250 統計的手法
TQMを勧めていく上で重要な要素
第2日目 セミナー内容
820.70 生産及び工程管理
工程管理方法と工程検V&V
工程のリスクマネジメント(PFMEA)
820.72 検査・測定及び試験装置
意図した用途への適合性、測定器の精度管理とは
820.75 工程の妥当性確認
査察のキーポイントであり、TQMが目指す試験レスとは
バリデーションの手法( VMP,DQ、IQ、OQ、PQ )
820.198 苦情ファイル
査察のキーポイント、苦情の管理の目的を理解する
820.100 是正処置及び予防措置
修正、是正、予防の相違とCAPA7ステップを理解する
CAPAの効果的なマネジメントと再発防止
FDA設計管理ガイダンスコース Rev.8
〜 設計管理規制だけではなく会社にとって利益をもたらすものづくりの本質 〜
満席につき募集を終了致しました!
次回のご出席をお待ちしております。
開催概要
新たに品質工学( タグチメソッド )、ロバスト設計を取り入れ、よりTQMにフォーカス
日 時:2011年11月30日(水) 10:00 〜 16:30
会 場:東京都立産業貿易センター浜松町館( 東京・浜松町 )
( JR浜松町駅から徒歩5分 )
対 象:医療機器・IVD市場にソフトウェア搭載機器でこれから参入しようと考えている企業
既に医療機器・IVDを海外に輸出している企業
FDA査察準備を考えている企業
受講料: ¥48,000( テキスト・規制邦訳文・昼食代含む )
割 引: ¥38,000( テキスト・規制邦訳文・昼食代含む )
1社2名以上ご参加の場合、ソフトウェアバリデーションコース(10月31日)
QSRコース(11月24,25日)を受講された方
特 割: すでに多くの方が受講されている企業様、オンサイトセミナーの後、継続研修などで
ご利用の場合、特別割引を適用させて頂きますのでご相談下さい。
定 員: 30名
( 出席者全員に教育記録として利用できる修了書をお渡ししています )
お申込: セミナー申し込みページから必要事項をご記入の上お申し込み下さい。
折り返し、会場、お振込先情報をメールでご連絡致します。
複数人ご参加される方はその旨ご記入下さい。 割引料金でご案内致します。
お問い合わせ: お問い合わせのページからお問い合わせ下さい。
お電話の方は下記セミナー事務局までお問い合わせ下さい。
042−856−2208
セミナー内容
1.設計品質の本質的価値
繰り返される設計品質に起因する製品不良
FDAが望んでいるTQMの本質、品質工学に基づくロバスト設計
医療機器の設計手法( Water Fall Model )
一貫性を問うFDA査察
〜 TQM、FDA設計管理ガイダンスに基づくQSRサブパートCの実践 〜
2.820.30 ( a ) 総則
TQMに欠かせないマネジメントチームの重要な役割
要員教育重要性( 820.25 )
3.820.30 ( b ) 設計及び開発計画
医療機器開発ライフサイクルマネジメント
タグチメソッドの開発方式による二段階設計法
PM,PLの役割
カスタマーニーズを要求仕様に変換する品質機能展開
コンカレントエンジニアリング
4.820.30 ( c ) 設計インプット
ロバスト設計による未然防止設計
要求仕様書から開発仕様書への変換
曖昧なニーズのスペックへの変換
トレーサビリティーマトリックス
5.820.30 ( d ) 設計アウトプット
入力のバラツキを押さえたアウトプットの確認
設計アウトプットを定義し文書類を明確にする
製造仕様書とは
DMRとのトレーサビリティー
6.820.30 ( e ) 設計審査
問題の早期発見、解決が目的
設計審査で設計のロバストネスを確認
審査者がキー
7.820.30 ( f ) 設計検証
設計検証と設計バリデーションの相違
インプットからアウトプットまでトレーサブルであるとは
設計検証レポートは、設計者の活動記録
ソフトウェアバリデーションとVモデル
3種類のバリデーションの相違
8.820.30 ( g ) 設計バリデーション
設計バリデーションの手法
ソフトウェアバリデーションとVモデル
3種類のバリデーションの相違
9.820.30 ( g ) 設計移管
設計部門が製造工程を設計する
工程V&VとQC工程表
機能展開とPFMEA
製造仕様書への展開
DHF,DMR,DHR
10.820.30 ( h ) 設計変更
重要な査察ポイント
設計変更プロセスとリスクアセスメント
コミュニケーションエラー
設計CAPA
医療機器・部材の開発に求められる品質の勘所
〜 「トレーサビリティー」と「設計根拠」を確立する 〜
このコースは【 日経ものづくり 】主催で開催されます。
http://techon.nikkeibp.co.jp/article/SEMINAR/20110809/195111/

開催概要
日 時:2011年9月26日(金) 10:00 〜 17:00 終了しました!
会 場:Learning Square 新橋( 東京・新橋 )
主 催:日経ものづくり
講 師;クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象:医療機器・IVD市場にすでに参入している企業、
これから参入を予定している企業の設計・生産技術、品質に関わる方
申 込:直接主催者HPからお申し込み下さい。
セミナー内容:
1.医療機器に求められる品質条件とは
ISOだけで品質を管理できると考えるのは大きな勘違いです。まず、人の命に直結する医療機器開発の本質について、なぜISOでは管理できないのか、一般的な製造業と照らし合わせながら説明します。
2.「 トレーサビリティー 」とは
医療機器では、製品の品質について顧客はもちろん第三者に対しても、顧客ニーズからはじまる設計インプットがどのように設計アウトプットや製造仕様書に落とし込まれたか、客観的に証明する必要があります。つまり、全ての手順が「追跡」可能でなければなりません。そのため、開発はウオーターフォール・モデルで行い、ライフサイクル全体で管理することになります。こうした考え方について解説します。
3.「 トレーサビリティー 」を確保するために必要なこと
まず、曖昧な顧客ニーズをどのように要求仕様書に変換し、開発者が理解できる開発仕様書に変換するのか、設計品質を作り込む上で最も重要なこの最初のプロセスの説明から、ほとんどの企業で理解されていない設計検証と設計バリデーションの相違を理解した上で、FDAが推奨する「トレーサビリティーマトリックス」の実践的な使用方法を紹介します。さらに設計のアウトプットである開発部門の責任で行う工程設計にどのように展開していくのか解説します。
4.「 設計根拠 」とは
医療機器開発における全ての活動は、客観的証拠(Objective Evidence)に基づいていなければなりません。これを支えるのが、ベリファイケーション(検証)およびバリデーション(妥当性確認)です。ところが、この2つをきちんと実行していなかったり、混同していたりする企業が少なくありません。医療機器開発で求められるエビデンスとは何かについて解説します。
5.「 設計根拠 」を確保するために必要なこと
客観的証拠(Objective Evidence)を確保するためには、設計検証の結果などを記録(DHF)として残し、トレーサビリティーマトリックスとリンクさせることです。しかし、この記録に残すという作業が日本の製造業の弱点でもあり、根拠を客観的証拠に基づいて示せない原因でもあります。まず、「記録」の概念を理解しすぐに実践するための例を挙げながら説明し、トレーサビリティーマトリックスと記録のリンクの重要性について解説します。

CAPA( 是正及び予防処置 )の実践
〜 品質不良の根本原因の究明・再発防止 〜
このコースは【 技術情報協会 】主催で開催されます。
http://www.gijutu.co.jp/doc/s_109108.htm
開催概要
日 時:2011年9月16日(金) 12:30 〜 16:30
会 場:大田区産業プラザ 6F C会議室( 東京・大田区 )
主 催:技術情報協会
講 師;クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に参入しているが医療機器を想定した開発体制が未整備な企業
米国に医療機器を輸出していてFDA査察準備をしている企業
医療機器に限らず品質問題撲滅のための方法を模索している企業
ISOでは品質問題が解決しないと嘆いている企業
FDA 査察で Form 483 で指摘をされレスポンスをしなければならない企業
申 込:直接主催者HPからお申し込み下さい。 こちらから。
講師割引:以前弊社オープンセミナー、オンサイトセミナーに御参加頂いた企業様は割引がございます。
お申し込みは開催日とコース名講師割引希望とご記入の上こちらからお申し込み下さい。
セミナー内容:
1.導入
a. 患者の命と会社を守る為に
b. Quality Cost 品質をお金で表現
c. 行政によるミッションの相違
d. 米国と日本の行政の相違
2.医療機器規制を理解する
a. QSRとISOの相違
b. ガイダンスの理解
c. QSIT を理解する
d. Warning Letter から理解できること
e. 日本の企業が受けた Warning Letter から理解できること
f. 要求規制とガイダンス
g. 規制を正しく理解するために
h. 規制・規格の用語を国際用語に揃える
3.再発する品質不良
a. なぜ品質不良は再発するのか?
b. FDA Expert の分析
4.用語の正しい理解
a. 3つの用語を理解する
b. 修正、是正処置、予防処置
c. 修正と是正の違い
d. 是正と予防の違い
5.規制要求事項
a. 21CFR820.100
b. 現存 or 潜在する問題の抽出
c. 再発問題の抽出
d. Warning Letter Case Study
6.CAPA7ステップ
Step 1. 問題を検出し明確にする
Step 2. 問題を詳しく調査する
Step 3. 根本原因を特定する
Step 4. 是正又は予防するための処置
Step 5. 処置の結果を文書化
Step 6. 処置の検証 or 妥当性確認を実施
Step 7. 有効性の確認
7.日常の対処方法
a. 簡易CAPAを使う
b. ディシジョンツリー
c. 潜在的問題の抽出と予防処置
8.CAPA のマネジメント
a. 品質問題は管理する
b. Warning Letter Case Study
9.事例研究
FDA入門コース3
〜 FDA査察に備える・医療機器開発の本質 〜
このコースは【 株式会社情報機構 】主催で開催されます。
http://www.johokiko.co.jp/seminar_medical/AA110847.php
開催概要
日 時:2011年8月26日(金) 12:30 〜 16:30
会 場:東京都江東区産業会館 2F第一会議室( 東京・東陽町 )
主 催:株式会社情報機構
講 師;クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に参入しているが医療機器を想定した開発体制が未整備な企業
米国に医療機器を輸出していてFDA査察準備をしている企業
申 込:直接主催者HPからお申し込み下さい。 こちらから。
講師割引:講師割引がございます。
お申し込みは開催日とコース名講師割引希望とご記入の上こちらからお申し込み下さい。
セミナー内容:
1.ISOだけではFDA査察にパスしない理由
規制をリスクとして考えないマネジメント
ISOで品質管理ができているという大きな勘違い
2.警告書( Warning Letter )から見えてくる査察の目線
警告書を統計的に理解するとわかる事実
警告書の内容とウェブ掲載の効果
3.QSRを理解するためのガイドライン
ガイドラインを理解しないでQSRは理解できない
間違った解釈をしない為に
4.QSITから理解すべきこと
QSITから見えてくるFDAの視線
人の命がかかる医療機器開発の本質を理解する
5.一番指摘が多く、まったく日本の企業に理解されていないCAPA
修正とCAとPAの違い
品質問題が再発するCAPA手順
6.製品実現のトレーサビリティーとは?
ウォーターフォールモデルで実証すること
ライフサイクで管理することの意味
7.根拠に基づくエビデンスで保証される活動とは?
全ての活動が根拠に基づくエビデンスで保証されなければならない
まったく理解されていない検証とバリデーション
8.リスクアセスメント
ライフサイクル全体で実施すべきリスクマネジメント
リスクアセスメントから導かれる製品、工程への展開
8.FDA査察の実際
FDAの組織と米国の法規制
査察の種類と頻度
9.FDA査察準備プログラム
査察対応プロジェクトを推進するには?
医療機器開発の本質を理解している者が必要
FDA CAPA( 是正及び予防処置 )
〜 査察で最も指摘の多いCAPAの正しい理解と問題を再発させない仕組み 〜
このコースは【 株式会社情報機構 】主催で開催されます。
http://www.johokiko.co.jp/seminar_medical/AA110670.php
開催概要
日 時:2011年6月17日(金) 12:30 〜 16:30
会 場:タワーホール船堀 4F401会議室( 東京・船堀 ) 部屋が変更されました!
主 催:株式会社情報機構
講 師;クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に参入しているが医療機器を想定した開発体制が未整備な企業
米国に医療機器を輸出していてFDA査察準備をしている企業
医療機器に限らず品質問題撲滅のための方法を模索している企業
ISOでは品質問題が解決しないと嘆いている企業
FDA 査察で Form 483 で指摘をされレスポンスをしなければならない企業
申 込:直接主催者HPからお申し込み下さい。 こちらから。
講師割引:講師割引がございます。以前弊社セミナー、オンサイトセミナーに御参加頂いた企業様は
割引がございます。誰が参加したのかご不明の場合でもこちらで確認できます。
お申し込みは開催日とコース名講師割引希望とご記入の上こちらからお申し込み下さい。
セミナー内容:
1.導入
a. 患者の命と会社を守る為に
b. Quality Cosit 品質をお金で表現
c. 行政によるミッションの相違
d. 米国と日本の行政の相違
2.医療機器規制を理解する
a. QSRとISOの相違
b. ガイダンスの理解
c. QSIT を理解する
d. Warining Letter から理解できること
e. 日本の Warining Letter から理解できること
f. 要求規制とガイダンス
g. 規制を正しく理解するために
h. 規制・規格の用語を国際用語に揃える
3.再発する品質不良
a. なぜ品質不良は再発するのか?
b. FDA Expert の分析
4.用語の正しい理解
a. 3つの用語を理解する
b. 修正、是正処置、予防処置
c. 修正と是正の違い
d. 是正と予防の違い
5.規制要求事項
a. 21CFR820.100
b. 現存 or 潜在する問題の抽出
c. 再発問題の抽出
d. Warning Letter Case Study
6.CAPA7ステップ
Step 1. 問題を検出し明確にする
Step 2. 問題を詳しく調査する
Step 3. 根本原因を特定する
Step 4. 是正又は予防するための処置
Step 5. 処置の結果を文書化
Step 6. 処置の検証 or 妥当性確認を実施
Step 7. 有効性の確認
7.日常の対処方法
a. 簡易CAPAを使う
b. ディシジョンツリー
c. 潜在的問題の抽出と予防処置
8.CAPA のマネジメント
a. 品質問題は管理する
b. Warning Letter Case Study
9.事例研究
医療機器に求められる品質要件★徹底解説
〜 Electronic Journal 第786回 Technical Seminar 〜
このコースは【 株式会社電子ジャーナル 】主催で開催されます。
http://www.electronicjournal.co.jp/
開催概要
日 時:2011年6月7日(火) 10:00 〜 17:00
会 場:総評会館( 東京・お茶の水 )
主 催:株式会社電子ジャーナル
講 師;クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象:医療機器・IVD市場にこれから参入しようと考えている企業
既に参入しているが医療機器を想定した開発体制が未整備な企業
米国に医療機器を輸出していてFDA査察準備をしている企業
申 込:直接主催者HPからお申し込み下さい。
講師割引:HPからお申し込み頂く際に「備考欄」でクオリス・イノーバのHPを見た
とお書き頂くと割引が適用されます。
定 員:40名
セミナー内容:
1.ISOマインドと医療機器製造
2.米国FDA規制・ガイドラインと医療機器開発のシステム構築
3.ライフサイクルマイルストーン、トレーサビリティーマトリックス、リスクアセスメントの概念
4.ISOの盲点と管理システムの構築
5.設計検証と設計バリデーションの違い
6.説明責任の順守と患者/企業を守る設計製造記録
7.工程バリデーションとCSV
8.CAPAの本質
9.社内準備のコンセンサス作り


ISO13485とFDA QSR( Quality System Regulation )の違いの中で、最も明確に異なるのは設計管理と言っても過言ではありません。 クオリス・イノーバがFDA査察準備プログラムで実施している模擬監査においても設計管理は一様に出来ていないのが実態です。 FDAは多くの企業査察経験と不具合報告、リコールの解析から、品質不良の原因が設計管理にあることを統計的に理解しています。 FDA査察官向け査察ガイドライン( QSIT )の代表的な4項目のひつとして設計管理が取り上げられている事からも、FDAが査察で何にフォーカスしているのかが理解出来ます。 QSRを理解するためにもFDAの設計管理ガイダンスを理解する事が重要ですが、医療機器開発の本質をこのガイダンスから学ぶことができます。 しかし、よくまとめられたこのガイダンスも、今までのISOの仕組みになれている方には読んだだけでは実際にどうやって仕組みを構築し直せばよいのか判りません。 そこで、クオリス・イノーバでは、GEヘルスケアの製品開発手法DFSS( Design for Six Sigma )を例に挙げながら、医療機器開発のライフサイクル設計について実践的で分かり易い説明をしています。 開発エンジニアのみならず、製品企画担当のマーケティングの方や品質保証の方、工程設計をする生産技術の方にも参加して頂きたい価値あるコースです。
講 師: クオリス・イノーバ代表 木村 浩実
対 象: 以下の方々
これから医療機器を米国に輸出する計画がある
すでに医療機器を米国に輸出している
FDA査察準備を行う予定
セミナー内容
1.設計管理の本質的価値

設計管理の本質的価値を理解する
Water Fall Process
21CFR820 Subpart C Design Control
2.重要な品質要素 820.30(a) 総則
経営スタッフの重要な役割
要員の教育 820.25
教育の重要性
3.設計及び開発計画 820.30(b)
ライフサイクルマイルストーン
プロダクトマネジャーとプロダクトリーダーの役割
カスタマーニーズの精度を上げる事の重要性
カスタマーCTQ
カスタマーニーズの要求仕様書への変換
コンカレントエンジニアリング
4.設計インプット 820.30(c)
要求仕様書から開発仕様書への変換
曖昧なニーズの基準値への変換
トレーサビリティーマトリックスの理解
設計インプットが製品品質を決める
5.設計アウトプット 820.30(d)
設計アウトプットを定義し明確にする
製造仕様書とは
DMRとのトレーサビリティー
6.設計審査 820.30(e)
問題の早期発見、解決が目的
設計審査プラン
設計審査は重要な設計品質ポイント
7.設計検証 820.30(f)
検証と設計バリデーションの相違
全てがトレーサブルであるとは
設計検証レポートは開発者の活動記録
ソフトウェアバリデーションとVモデル
設計バリデーション、ソフトウェアバリデーション、
プロセスバリデーションのアプローチの違いを理解する
8.設計移管 820.30(h)
開発が行うべき設計移管の重要な要素
工程V&VとQC工程表
機能展開とプロセスリスクアセスメント
製造仕様書への変換
DHF、DMR、DHR
9.設計変更 820.30(i)
重要な査察ポイント
リスクアセスメントと変更プロセス
コミュニケーションエラー
設計CAPA
医療機器開発の勘所
〜 新規参入のための手引き 〜
このコースは 【 日経ものづくり・日経デジタルヘルス 】主催で実施致します。
http://techon.nikkeibp.co.jp/article/SEMINAR/20110112/188659/
開催概要
日 時: 2011年2月28日(月) 10:00〜17:00 終了しました!
会 場: 【東京・新橋】Leraning Square 新橋
http://www.learningsite21.com/ls4/e01.html
主 催: 日経ものづくり
講 師: クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象: *医療機器市場にこれから参入しようと考えている企業
*既に参入しているものの医療機器を想定した開発体制が未整備な企業
*品質に関する査察の厳しい地域(特に米国)に向けた医療機器を開発している企業・
これから開発しようとしている企業
*医療機器向けに部品やユニット、ソフトウェア、ITシステムなどを供給している企業
申込み: 外部セミナーハウスに直接お申し込み下さい。
http://techon.nikkeibp.co.jp/article/SEMINAR/20110112/188659/
セミナー内容:
医療機器に求められる品質条件とは
ISOだけで品質を管理できると考えるのは大きな勘違いです。初めに、人の命に直結する医療機器開発の本質について、一般的な製造業と照らし合わせながら説明します。
医療機器に関連する規制、国際規格、査察
医用機器にはどのような規制や国際規格があるのか、概要を紹介します。また、医療機器には政府機関による査察という制度が存在します。ここでは、那珂でも特に重要な米国食品医薬品局(FDA)による査察について最新の情勢を紹介します。
製品開発の「トレーサビリティー」
医療機器では、製品の品質について顧客はもちろん第三者に対しても客観的に説明する必要があります。つまり、全ての手順、データが「追跡」可能でなければなりません。そのため、開発はウォーターフォールモデルで開発を行い、ライフサイクル全体で管理することになります。こうした考え方について解説します。
「根拠」に基づく意志決定と設計
医療機器開発における全ての活動は、客観的な「根拠(エビデンス}」に基づいていなければなりません。これを支えるのが、バリデーション(妥当性確認)とベリフィケーション(検証)です。ところが、この2つをきちんと実行していなかったり、混同していたりする企業が少なくありません。医療機器開発で求められるエビデンスとは何か?そのために何をしなければならないのかについて説明します。
事例による理解度チェック
どれだけ知識を習得しても、それを現場で実践できなければ意味がありません。豊富な事例に基づき、学んだ知識を製品開発の場でどう活かせばよいのかについて理解を深めて頂きます。

医療機器・体外診断薬のためのFDA入門コース
〜 FDA査察に備える・医療機器開発の本質 〜
このコースは外部セミナーハウスで実施致します。
http://www.johokiko.co.jp/seminar_medical/AA110271.php
開催概要
日 時: 2011年2月16日(水) 12:30〜16:30
会 場: 【東京・船堀】タワーホール船堀3階306会議室
http://www.towerhall.jp/4access/access.html
主 催: 情報機構
講 師: クオリス・イノーバ株式会社 代表取締役社長 木村 浩実
対 象: 医療機器・体外診断薬を米国に輸出しようと考えておられる企業
既に輸出をしていてFDAの査察準備の為の初歩的な知識を学びたい方
ISOと何が異なるのかFDAの規制を理解する手掛かりとしたい方
申込み: 外部セミナーハウスに直接お申し込み下さい。
http://www.johokiko.co.jp/mousikomi/index.php#no1
以前クオリス・イノーバ主催の講習会に参加された企業の方で割引を希望される方は、
こちらから過去に参加された日と割引希望と記入の上ご連絡頂ければ、割引適用の申込
用紙をお送りいたします。
セミナー内容:
1.ISOだけではFDA査察にパスしない理由
規制をリスクとして考えないマネジメント
ISOで品質管理ができているという大きな勘違い
2.警告書( Warning Letter )から見えてくる査察の目線
警告書を統計的に理解するとわかる事実
警告書の内容とウェブ掲載の効果
3.QSRを理解するためのガイドライン
ガイドラインを理解しないでQSRは理解できない
間違った解釈をしない為に
4.QSITから理解すべきこと
QSITから見えてくるFDAの視線
人の命がかかる医療機器開発の本質を理解する
5.一番指摘が多く、まったく日本の企業に理解されていないCAPA
修正とCAとPAの違い
品質問題が再発するCAPA手順
6.製品実現のトレーサビリティーとは?
ウォーターフォールモデルで実証すること
ライフサイクで管理することの意味
7.根拠に基づくエビデンスで保証される活動とは?
全ての活動が根拠に基づくエビデンスで保証されなければならない
まったく理解されていない検証とバリデーション
8.リスクアセスメント
ライフサイクル全体で実施すべきリスクマネジメント
リスクアセスメントから導かれる製品、工程への展開
9.FDA査察の実際
FDAの組織と米国の法規制
査察の種類と頻度
10.FDA査察準備プログラム
査察対応プロジェクトを推進するには?
医療機器開発の本質を理解している者が必要
2011年セミナーのご案内
各コースの概要についてはFDAセミナーのページを、コースの詳細はこのページに掲載される開催詳細を御覧下さい。 下記の開催予定表に予定日が確定し、このページに開催の詳細が掲載されたコースはお申し込みが可能です。 予定表に開催予定日が確定していないコースのお申し込み、受講料等の詳細は、約1ヶ月半前にこのページでお知らせいたします。 その後セミナー申込みページからお申し込み下さい。 セミナーや、オンサイトセミナーについてのお問い合せはこちらからお願いします。
* お客様のFDA査察サポートの為予定通りセミナーが実施できない場合がございます。 予めご了解下さい。 予定表に日付が表示されるとセミナーは確定となり、白丸は外部セミナーハウスでの開催となり、このページでもご紹介します。( 講師割引があるのでご利用下さい )
( 2011 Mar. 22 update )
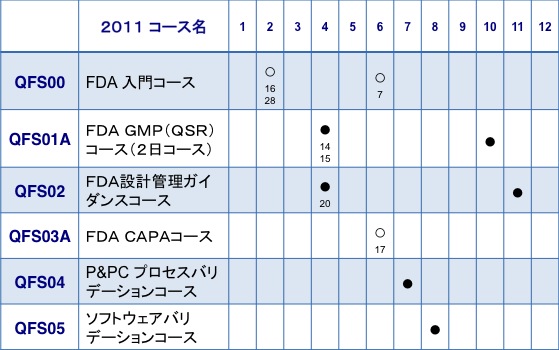
お待たせしました。 今年最後のQSRトレーニングコースとなります。
医療機器・IVD を米国に輸出している、或いは輸出を考えている会社にとって、FDAのGMPは品質システムとして適合させなければならない必要条件です。 また、ISO13485だけの品質システムではFDAの厳しい査察にパスしないのが現実です。 クオリス・イノーバGAP監査結果の数字( 平均35% )を見てもISOだけでは無理があることがおわかりいただけると思います。 しかし、FDAGMPであるQSR( Quality System Regulation )とISOを比較しても、その違いは読んだだけでは理解できません。 QSRを理解するには、規制の序文( Preamble )、FDA査察官向け査察ガイド( QSIT )、さまざまなガイドラインを理解することが重要です。 本コースでは、多くの企業監査、FDA査察の経験とFDAの企業への最新の警告書( Warning Letter )から要求事項の本質をわかりやすく説明するコースです。 全世界に100以上の工場を持つGEヘルスケアのコーポレートオーディターとして全世界の工場を監査してきた豊富な実績とFDA査察経験、GEシックスシグマ・ブラックベルトとして多くの品質改善を経験してきた実績で、規制の要求事項のみならず医療機器開発の本質を鋭く説く実践コースです。
開催概要
お申込み受付を終了致しました!
日 時: 2010年11月5日( 金曜日 )10:00〜16:30

会 場: 東京都立産業貿易センター 浜松町館
東京都港区海岸1−7−8
( JR浜松町駅から徒歩5分 )
講 師: クオリス・イノーバ代表 木村 浩実
対 象: QSRの基礎を学び導入する、
或いはFDA査察準備予定の方
受講料: ¥47,000( テキスト・規制邦訳文・昼食代含む )
割 引: ¥39,000( テキスト・規制法訳文・昼食代含む )
以前弊社セミナーにご参加された方、
続けて複数コース受講される方、
1社2名以上ご参加される方
聴 講: ¥9,000( テキスト・昼食代含む )
以前本コースを受講された方でもう一度
受講して理解を深めたい方
( 席が空いている場合に限らせて頂きます )
特 割: すでに多くの方が受講されている企業様、継続研修などでご利用の場合、特別割引を適用させて
頂きますのでご相談下さい。 NEW!
定 員: 30名( お早めにお申し込み下さい )
申込み: セミナー申込みページから必要事項を記入しお申し込み下さい。
折り返し、会場、お振込先の情報をメールでご連絡致します。
複数人ご参加、複数コース受講、以前受講された方は、その旨ご記入下さい。
また、聴講ご希望の方は、聴講希望と記載され、以前受講された日付をご記入下さい。
FAXでのお申し込みをご希望の方は、セミナー申込み画面をコピーし、必要事項をご記入の上、
042-856-2210まで送信下さい。
お問い合せ: お問い合せのページから必要事項をご記入の上お問い合せ下さい。
メールの方はこちらのアドレスから、お電話の方は下記までお問い合せ下さい。
セミナー事務局 mail@qualysinnova.com 042-856-2208
申込締切: 10月28日(木曜日)
* 教育記録として利用できる修了書を受講者様に発行しております。
セミナー内容
1.FDA規制の本質

FDAのミッション
FDAの組織
米国の法規制
ISOとQSRの本質的な違い
理解すべきガイダンス
ISOと品質管理の相違
品質管理の本質を理解する
FDA査察とは
情報の入手
2.FDA査察の本質
FDA査察とは
査察の頻度
品質不良が招く査察リスク
Warning Letter
FDA査察対応準備
査察の本質
3.QSITとは
QSIT手法とは
QSITアプローチ
FDA解析データ
4つのフォーカスエリア
4.規制要求事項の実践的
820.20 経営者の責任
マネジメントレビューの内容
820.22 品質監査
監査の視点と報告書
820.25 要員
力量表とアウェアネストレーニング
820.30 設計管理
ライフサイクルマイルストーン開発
トレーサビリティーマトリックス
設計検証と設計バリデーション
820.40 文書管理
フォーマットの作成
820.50 購買管理
これからの潮流、サプライヤー監査
820.60 識別
識別の手法
820.65 トレーサビリティー
リコール時に役立つトレース
820.70 生産及び工程管理
工程検証と工程バリデーション
820.72 検査、測定及び試験装置
意図した用途に適切とは
820.75 工程の妥当性確認( プロセスバリデーション )
DQ,IQ、OQ、PQ
820.80 受領、工程内及び完成機器の受入
完成機器の受入とは
820.86 受入の状態
状態表示の方法
820.90 不適合品
不適合品を管理するとは
820.100 是正処置及び予防措置( CAPA )
修正、是正、予防の違いを理解する
820.120 機器のラベリング
ラベリングの管理方法
820.130 機器の包装
包装設計は開発の仕事
820.140 取扱い
取扱マークとは
820.150 保管
ペストコントロール
820.160 流通
流通記録とは
820.170 据付
インスタレーションマニュアル
820.180 記録・一般要求事項
保管期間は?
820.181 機器原簿( DMR )
820.184 機器履歴簿( DHR )
820.186 品質システム記録
820.198 苦情ファイル
苦情情報の取扱と管理
820.200 付帯サービス
MDRとマネジメントへの報告
820.250 統計的手
品質改善に欠かせない統計的手法
今年最後の設計管理ガイダンスコースです。
ISO13485とFDA QSR( Quality System Regulation )の違いの中で最も明確に異なるのは設計管理と言っても過言ではありません。 FDAは多くの企業査察の経験と不具合報告、リコールの解析から、品質不良の原因が設計管理にあることを統計的に理解しており、その根拠としてFDA査察官向け査察ガイドラインの4項目のひつとして設計管理を挙げています。 また、FDAは設計管理ガイダンスを出していますが、このガイドラインを理解することがQSRを理解することであり、医療機器・IVD開発の本質を学ぶことになります。 しかし、よくまとめられたこのガイダンスでも読んだだけでは実践に結びつきません。 そこで、クオリス・イノーバでは、GEヘルスケアの製品開発手法DFSS( Design for Six Sigma )を例に挙げ、医療機器開発のライフサイクル設計について分かり易く説明をしています。 医療機器開発全般の理解を深める開発エンジニアのみならず、製品企画担当のマーケティング部門の方にも出席して頂きたい価値ある人気のコースです。
開催概要
お申し込み受付を終了致しました!
日 時: 2010年11月29日( 月曜日 )10:00〜16:30
会 場: 東京都立産業貿易センター 浜松町館
東京都港区海岸1−7−8
( JR浜松町駅から徒歩5分 )
講 師: クオリス・イノーバ代表 木村 浩実
対 象: 設計管理の基礎、開発手法の理解を深め
医療機器開発の本質を学びたい方
或いはFDA査察準備をする方
製品企画、マーケティングの方
受講料: ¥47,000( テキスト・邦訳・昼食代含む )
割 引: ¥39,000( テキスト・邦訳・昼食代含む )
以前弊社セミナーにご参加された方、
続けて複数コース受講される方、
1社2名以上ご参加される方
聴 講: ¥9,000( テキスト・昼食代含む )
以前本コースを受講された方でもう一度
受講して理解を深めたい方
( 席が空いている場合に限らせて頂きます )
特 割: すでに多くの方が受講されている企業様、継続研修などでご利用の場合、
特別割引を適用さて頂きますのでご相談下さい。 NEW!
定 員: 30名( お早めにお申し込み下さい )
申込み: セミナー申込みページから必要事項を記入しお申し込み下さい。
折り返し、会場、お振込先の情報をメールでご連絡致します。
複数人ご参加、複数コース受講、以前受講された方は、その旨ご記入下さい。
また、聴講ご希望の方は、聴講希望と記載され、以前受講された日付をご記入下さい。
FAXでのお申し込みをご希望の方は、セミナー申込み画面をコピーし、必要事項をご記入の上、
042-856-2210まで送信下さい。
お問い合せ: お問い合せのページから必要事項をご記入の上お問い合せ下さい。
メールの方はこちらのアドレスから、お電話の方は下記までお問い合せ下さい。
セミナー事務局 mail@qualysinnova.com 042-856-2208
申込締切: 11月24日(水曜日)
* 教育記録として利用できる修了書を受講者に発行しております。
セミナー内容
1.設計管理の本質的価値
 設計管理の本質的価値を理解する
設計管理の本質的価値を理解する
Water Fall Process
21CFR Subpart C Design Control
2.重要な品質要素 820.30(a) 総則
経営スタッフの重要な役割
要員の教育 820.25
教育の重要性
3.設計及び開発計画 820.30(b)
ライフサイクルマイルストーン
カスタマージーズの精度を上げる
カスタマーCTQ
ニーズのエンジニアリング仕様への変換
コンカレントエンジニアリング
4.設計インプット 820.30(c)
設計管理で最も重要なフェーズ
曖昧なニーズの基準値への変換
トレーサビリティーマトリックスの理解
設計インプットが製品品質を決める
5.設計アウトプット 820.30(d)
設計アウトプットを定義し明確にする
DMRとのトレーサビリティー
6.設計審査 820.30(e)
問題の早期発見、解決が目的
設計審査プラン
設計審査は重要な設計品質ポイント
7.設計検証 820.30(f)
検証と設計バリデーションの相違
全てがトレーサブルとは
設計検証レポートは開発者の活動記録
ソフトウェアバリデーションとVモデル
設計バリデーション、ソフトウェアバリデーション、
プロセスバリデーションのアプローチの違いを理解する
8.設計移管 820.30(h)
設計移管の重要な要素
機能展開とプロセスリスクアセスメント
DHF、DMR、DHR
9.設計変更 820.30(i)
設計変更管理手順
コミュニケーションエラー
重要な査察ポイント
このコースは外部セミナーハウスによる開催となります。
FDAの査察手法QSITの4つの査察重要項目の一つであるP&PC( Production & Process Controls :生産及び工程管理 )の内、最も指摘の多い自動化工程、プロセスバリデーションにフォーカスし、QSRの要求事項( 21CFR Part 820 )のみならず、QSIT( FDA査察官向け査察ガイド )、FDAガイドライン、DHTFガイドライン、医薬品GMPなどさまざまな角度から要求事項の本質を紐解き、製造工程を科学的根拠をもって設計し検証するプロセスを学びます。 CSV( Cpmputerized System Validation )の根拠となる自動化工程の識別やバリデーションの基礎についても言及。 FDAの規制準拠のみならず、製造工程の検証・妥当性確認の本質を学ぶ、医療機器・体外診断薬製品の開発・製造・品質保証部門の方に最適なコースです。
開催概要
終了しました!
日 時: 2010年10月22日( 金曜日 )10:30〜16:00
このコースは外部セミナーハウスによる開催となります。 詳しくはこちらを御覧下さい。
http://www.johokiko.co.jp/seminar_medical/AA101072.php
医療機器・IVD を米国に輸出している、或いは輸出を考えている会社にとって、FDAのGMPは品質システムとして適合させなければならない必要条件です。 また、ISO13485だけの品質システムではFDAの厳しい査察にパスしないのが現実です。 クオリス・イノーバGAP監査結果の数字( 平均35% )を見てもISOだけでは無理があることがおわかりいただけると思います。 しかし、FDAGMPであるQSR( Quality System Regulation )とISOを比較しても、その違いは読んだだけでは理解できません。 QSRを理解するには、規制の序文( Preamble )、FDA査察官向け査察ガイド( QSIT )、さまざまなガイドラインを理解することが重要です。 本コースでは、多くの企業監査、FDA査察の経験とFDAの企業への最新の警告書( Warning Letter )から要求事項の本質をわかりやすく説明するコースです。 全世界に100以上の工場を持つGEヘルスケアのコーポレートオーディターとして全世界の工場を監査してきた豊富な実績とFDA査察経験、GEシックスシグマ・ブラックベルトとして多くの品質改善を経験してきた実績で、規制の要求事項のみならず医療機器開発の本質を鋭く説く実践コースです。
開催概要
日 時: 2010年4月5日( 月曜日 )10:00〜16:30
会 場: 東京都立産業貿易センター 浜松町館
東京都港区海岸1−7−8
( JR浜松町駅から徒歩5分 )
講 師: クオリス・イノーバ代表 木村 浩実
対 象: QSRの基礎を学び導入する、
或いは査察準備予定の方
受講料: ¥47,000( テキスト・昼食代含む )
割 引: ¥40,000( テキスト・昼食代含む )
以前弊社セミナーにご参加された方、
続けて複数コース受講される方、
1社2名以上ご参加される方
聴 講: ¥9,000( テキスト・昼食代含む )NEW!
以前本コースを受講された方でもう一度
受講して理解を深めたい方
( 席が空いている場合に限らせて頂きます )
定 員: 30名( お早めにお申し込み下さい )
申込み: セミナー申込みページから必要事項を記入しお申し込み下さい。
折り返し、会場、お振込先の情報をメールでご連絡致します。
複数人ご参加、複数コース受講、以前受講された方は、その旨ご記入下さい。
また、聴講ご希望の方は、聴講希望と記載され、以前受講された日付をご記入下さい。
FAXでのお申し込みをご希望の方は、セミナー申込み画面をコピーし、必要事項をご記入の上、
042-856-2210まで送信下さい。
お問い合せ: お問い合せのページから必要事項をご記入の上お問い合せ下さい。
メールの方はこちらのアドレスから、お電話の方は下記までお問い合せ下さい。
セミナー事務局 mail@qualysinnova.com 042-856-2208
申込締切: 3月30日(火曜日)
* 教育記録として利用できる修了書を受講者に発行しております。
セミナー内容
1.FDA規制の本質
FDAのミッション
FDAの組織
米国の法規制
ISOとQSRの本質的な違い
理解すべきガイダンス
情報の入手
2.FDA査察の本質
FDA査察とは
査察の頻度
品質不良が招く査察リスク
Warning Letter
FDA査察対応準備
査察の本質
3.QSITとは
QSIT手法とは
QSITアプローチ
FDA解析データ
4つのフォーカスエリア
4.規制要求事項の実践的
820.20 経営者の責任
820.22 品質監査
820.25 要員
820.30 設計管理
820.40 文書管理
820.50 購買管理
820.60 識別
820.65 トレーサビリティー
820.70 生産及び工程管理
820.72 検査、測定及び試験装置
820.75 工程の妥当性確認( プロセスバリデーション )
820.80 受領、工程内及び完成機器の受入
820.86 受入の状態
820.90 不適合品
820.100 是正処置及び予防措置( CAPA )
820.120 機器のラベリング
820.130 機器の包装
820.140 取扱い
820.150 保管
820.160 流通
820.170 据付
820.180 記録・一般要求事項
820.181 機器原簿( DMR )
820.184 機器履歴簿( DHR )
820.186 品質システム記録
820.198 苦情ファイル
820.200 付帯サービス
820.250 統計的手法
コース概要・受講者様の声
コースパンフレット( 現在準備中 )
ISO13485とFDA QSR( Quality System Regulation )の違いの中で最も明確に異なるのは設計管理と言っても過言ではありません。 FDAは多くの企業査察の経験と不具合報告、リコールの解析から、品質不良の原因が設計管理にあることを統計的に理解しており、その根拠としてFDA査察官向け査察ガイドラインの4項目のひつとして設計管理を挙げています。 また、FDAは設計管理ガイダンスを出していますが、このガイドラインを理解することがQSRを理解することであり、医療機器・IVD開発の本質を学ぶことになります。 しかし、よくまとめられたこのガイダンスでも読んだだけでは実践に結びつきません。 そこで、クオリス・イノーバでは、GEヘルスケアの製品開発手法DFSS( Design for Six Sigma )を例に挙げ、医療機器開発のライフサイクル設計について分かり易く説明をしています。 設計のみならず、医療機器開発全般の理解を深める開発エンジニアのみならず、製品企画担当のマーケティング部門の方にも出席して頂きたい価値ある人気のコースです。 ( ソフトウェアの開発基礎にも言及し、ソフトウェアバリデーションコースを受講の方は受講前に設計管理についてご理解ください。)
開催概要
日 時: 2010年4月6日( 火曜日 )10:00〜16:30
会 場: 東京都立産業貿易センター 浜松町館
東京都港区海岸1−7−8
( JR浜松町駅から徒歩5分 )
講 師: クオリス・イノーバ代表 木村 浩実
対 象: 設計管理の基礎、開発手法の理解を深め
医療機器開発の本質を学びたい方
或いはFDA査察準備をする方
製品企画、マーケティングの方
受講料: ¥47,000( テキスト・昼食代含む )
割 引: ¥40,000( テキスト・昼食代含む )
以前弊社セミナーにご参加された方、
続けて複数コース受講される方、
1社2名以上ご参加される方
聴 講: ¥9,000( テキスト・昼食代含む )NEW!
以前本コースを受講された方でもう一度
受講して理解を深めたい方
( 席が空いている場合に限らせて頂きます )
定 員: 30名( お早めにお申し込み下さい )
申込み: セミナー申込みページから必要事項を記入しお申し込み下さい。
折り返し、会場、お振込先の情報をメールでご連絡致します。
複数人ご参加、複数コース受講、以前受講された方は、その旨ご記入下さい。
また、聴講ご希望の方は、聴講希望と記載され、以前受講された日付をご記入下さい。
FAXでのお申し込みをご希望の方は、セミナー申込み画面をコピーし、必要事項をご記入の上、
042-856-2210まで送信下さい。
お問い合せ: お問い合せのページから必要事項をご記入の上お問い合せ下さい。
メールの方はこちらのアドレスから、お電話の方は下記までお問い合せ下さい。
セミナー事務局 mail@qualysinnova.com 042-856-2208
申込締切: 3月31日(水曜日)
* 教育記録として利用できる修了書を受講者に発行しております。
セミナー内容
1.設計管理の本質的価値
設計管理の本質的価値を理解する
コースパンフレット( 現在準備中 )
依然としてFDA査察で最も指摘の多いカテゴリーがCAPAであり、FDAはまず苦情報告からCAPAを査察することで、MDR( 不具合報告 )、CAR( リコール )が確実に報告されているかという法的順守を確認し、次にCAPAを見ることでその企業の品質改善の姿勢を理解しようとします。 しかし、ISOを取得している企業の場合、ほとんどと言っていいくらいMDRの要求事項やCAPAの本質を理解しておらず、CAPAで品質が改善されると思っていません。 FDAもこのことを査察やリコールの報告書などを通して統計的に理解しており、FDAの専門官はCAPAを理解している企業はほとんどないとまで言っています。 米国に輸出するしないに係わらず日本の医療機器開発メーカーはこれに気付くべきであり、CAPAの7ステップとその管理手法がシックスシグマ品質改善手法に似た手法であることを、GEヘルスケア、シックスシグマ・ブラックベルトの資格を持ち、多くの品質改善経験を手がけてきた講師が実践的な手法を説明し、CAPAを積極的に導入することによる価値に気付いて頂くDFA査察に十分対応したコースです。
開催概要
日 時: 2010年4月7日( 水曜日 )10:00〜16:00
会 場: 東京都立産業貿易センター 浜松町館
東京都港区海岸1−7−8
( JR浜松町駅から徒歩5分 )
講 師: クオリス・イノーバ代表 木村 浩実
対 象: QSRの基礎を学び導入する、
或いは査察準備予定の方
受講料: ¥45,000( テキスト・昼食代含む )
割 引: ¥38,000( テキスト・昼食代含む )
以前弊社セミナーにご参加された方、
続けて複数コース受講される方、
1社2名以上ご参加される方
聴 講: ¥7,000( テキスト・昼食代含む )NEW!
以前本コースを受講された方でもう一度
受講して理解を深めたい方
( 席が空いている場合に限らせて頂きます )
定 員: 30名( お早めにお申し込み下さい )
申込み: セミナー申込みページから必要事項を記入しお申し込み下さい。
折り返し、会場、お振込先の情報をメールでご連絡致します。
複数人ご参加、複数コース受講、以前受講された方は、その旨ご記入下さい。
また、聴講ご希望の方は、聴講希望と記載され、以前受講された日付をご記入下さい。
FAXでのお申し込みをご希望の方は、セミナー申込み画面をコピーし、必要事項をご記入の上、
042-856-2210まで送信下さい。
お問い合せ: お問い合せのページから必要事項をご記入の上お問い合せ下さい。
メールの方はこちらのアドレスから、お電話の方は下記までお問い合せ下さい。
セミナー事務局 mail@qualysinnova.com 042-856-2208
申込締切: 3月30日(火曜日)
* 教育記録として利用できる修了書を受講者に発行しております。
セミナー内容
1.査察の重要な要素CAPA
 FDAのミッション
FDAのミッションコースパンフレット( 現在準備中 )
FDAの査察手法QSITの4つの査察重要項目の一つであるP&PC( Production & Process Controls :生産及び工程管理 )の内、最も指摘の多い自動化工程、プロセスバリデーションにフォーカスし、QSRの要求事項( 21CFR Part 820 )のみならず、QSIT( FDA査察官向け査察ガイド )、FDAガイドライン、DHTFガイドライン、医薬品GMPなどさまざまな角度から要求事項の本質を紐解き、製造工程を科学的根拠をもって設計し検証するプロセスを学びます。 CSV( Cpmputerized System Validation )の根拠となる自動化工程の識別やバリデーションの基礎についても言及。 FDAの規制準拠のみならず、製造工程の検証・妥当性確認の本質を学ぶ、医療機器・体外診断薬製品の開発・製造・品質保証部門の方に最適なコースです。
開催概要
日 時: 2010年4月28日( 水曜日 )10:00〜16:30
会 場: 東京都立産業貿易センター 浜松町館
東京都港区海岸1−7−8
( JR浜松町駅から徒歩5分 )
講 師: クオリス・イノーバ代表 木村 浩実
対 象: QSRの基礎を学び導入する、
或いは査察準備予定の方
受講料: ¥47,000( テキスト・昼食代含む )
割 引: ¥40,000( テキスト・昼食代含む )
以前弊社セミナーにご参加された方、
続けて複数コース受講される方、
1社2名以上ご参加される方
聴 講: ¥9,000( テキスト・昼食代含む )NEW!
以前本コースを受講された方でもう一度
受講して理解を深めたい方
( 席が空いている場合に限らせて頂きます )
定 員: 30名( お早めにお申し込み下さい )
申込み: セミナー申込みページから必要事項を記入しお申し込み下さい。
折り返し、会場、お振込先の情報をメールでご連絡致します。
複数人ご参加、複数コース受講、以前受講された方は、その旨ご記入下さい。
また、聴講ご希望の方は、聴講希望と記載され、以前受講された日付をご記入下さい。
FAXでのお申し込みをご希望の方は、セミナー申込み画面をコピーし、必要事項をご記入の上、
042-856-2210まで送信下さい。
お問い合せ: お問い合せのページから必要事項をご記入の上お問い合せ下さい。
メールの方はこちらのアドレスから、お電話の方は下記までお問い合せ下さい。
セミナー事務局 mail@qualysinnova.com 042-856-2208
申込締切: 4月21日(水曜日)
* 教育記録として利用できる修了書を受講者に発行しております。
セミナー内容
1.FDAのミッション
 FDAとISOの相違
FDAとISOの相違
コースパンフレット( 現在準備中 )
* 開催延期のお知らせ *
お客様のFDA査察準備の為、5月28日開催予定のソフトウェアバリデーションコースは延期となりました。 皆様には大変ご迷惑をお掛け致しますが、ご理解を頂きますようお願い申し上げます。 次回開催は追ってご連絡を申し上げます。
医療機器のみならず、医用機器の開発、製造もソフトウェア無しには考えられないのが現実。 また、ソフトウェアの関与する不具合報告、リコールの増加からFDAもソフトウェアにフォーカスしてきているのも事実です。 本コースでは、FDA、QSRを根拠にバリデーションを要求される医療機器のソフトウェアバリデーション、自動化装置について、FDAガイドラインを中心に、IEC62304の要求事項も考慮しながらその本質を解説いたします。 ソフトウェア試験方法の箇所は、第三者検証事業をしておられる株式会社エス・キュー・シー様から専門的な立場から具体的な説明を行います。
開 催 概 要
日 時: 2010年5月28日( 金曜日 )10:00〜16:30
会 場: 東京都立産業貿易センター 浜松町館
東京都港区海岸1−7−8
( JR浜松町駅から徒歩5分 )
講 師: クオリス・イノーバ代表 木村 浩実
対 象: 医療機器ソフトウェア開発エンジニア
或いはFDA査察準備をする方
受講料: ¥47,000( テキスト・昼食代含む )
割 引: ¥40,000( テキスト・昼食代含む )
以前弊社セミナーにご参加された方、
続けて複数コース受講される方、
1社2名以上ご参加される方
定 員: 30名
申込み: セミナー申込みページから必要事項を記入しお申し込み下さい。
折り返し、会場、お振込先の情報をメールでご連絡致します。
複数人ご参加、複数コース受講、以前受講された方は、その旨ご記入下さい。
また、聴講ご希望の方は、聴講希望と記載され、以前受講された日付をご記入下さい。
FAXでのお申し込みをご希望の方は、セミナー申込み画面をコピーし、
必要事項をご記入の上、FAX番号042-856-2210まで送信下さい。
お問い合せ: お問い合せのページから必要事項をご記入の上お問い合せ下さい。
メールの方はこちらのアドレスから、お電話の方は下記までお問い合せ下さい。
セミナー事務局 mail@qualysinnova.com 042-856-2208
申込締切: 5月24日(月曜日)
* 教育記録として利用できる修了書を受講者に発行しております。
セミナー内容( 詳細は後日掲載 )
1.FDAガイダンスに基づく設計手法
2.ライフサイクル設計手法
3.トレーサビリティーマトリックスからの展開
4.設計審査
5.具体的なソフトウェア試験方法
( 株式会社エス・キュー・シー )
2010年セミナーのご案内
各コースの概要については、このページに掲載される開催概要を御覧下さい。 下記の開催予定表に予定日が確定し、このページに開催概要が掲載されたコースはお申し込みが可能です。 予定表に開催予定日が確定していないコースのお申し込み、受講料等の詳細は、約1ヶ月半前にこのページでお知らせいたします。 その後セミナー申込みページからお申し込み頂けます。 セミナーや、オンサイトセミナーについてのお問い合せはこちらからどうぞ。
* お客様のFDA査察サポートの為、予定通りセミナーが実施できない場合がございます。 予めご了解下さい。 予定表に日付が表示されるとセミナーは確定となり、白丸は外部セミナーハウスでの開催となります。
( 14 Sep. 2010 )
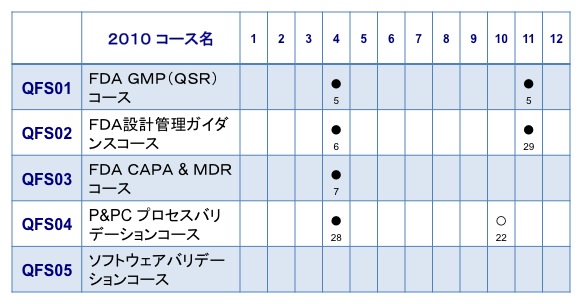
FDAの査察で依然として最も指摘が多いカテゴリーがCAPAであり、FDAは品質を改善するためのCAPA( 是正処置及び予防措置 )を見ることで、規制順守のみならず、企業の品質活動に対する姿勢を確認しようとします。そして、ISOでCAPAを理解しているはずの企業は、実はまったく理解していないとFDAは明言しています。このセミナーでは、FDAが実施したトレーニングから規制要求事項を説明し、FDAの Warning Letter などの事例からCAPA、MDR、CAR規制要求事項を理解します。FDAの規制に適合するフォーマットを実際に考え作り、セミナー終了後には、すぐに会社で使用できるCAPAフォーマットが完成することでしょう。クオリス・イノーバでは、100以上の工場を持つGEヘルスケアのコーポレートオーディターとして世界各国の工場を監査し、CAPAで改善させてきた実績と、GEシックスシグマ、ブラックベルトの資格を持ち、CAPAの7つの改善ステップがGEシックスシグマに似た品質改善活動である事を豊富な品質改善経験を通して説明し、日常的な活動としてCAPAを積極的に導入する方法を説明します。

日 時: 2009年6月16日(火曜)
10:00〜16:00
会 場: 東京都千代田区永田町: 薬業健保会館
講 師: クオリス・イノーバ 代表 木村 浩実
受講料: ¥47,000−( 資料・昼食込み )
¥40,000−( 1社2名様以上、或いは
以前弊社セミナーを受講された方 )
* 社内教育記録としてご活用用頂ける修了書を発行しております。
お申し込みは、セミナー申込み画面からお申し込み下さい。 折り返し、振込先、会場等のご案内を差し上げます。
申込締切:6月11日(木曜)
セミナーの内容
1.CAPAが査察の重要な要素である理由
FDA の解析と結果
2.CAとPAの相違
CAとPAの違いを理解する
CAとPAの例題
3.CAPAの規制要求事項
21CFR Part 820.100 CAPA
規制要求事項の重要ポイント
4.MDR、CARの規制要求事項
21CFR Part 803 MDR
21CFR Part 806 CAR
5.Warning Letter から学ぶ
同業者のWarning Letter から学ぶ
Warning Letter の検索方法
6.QSIT
CAPAはQSITの重要な要素
CAPAの適用範囲
7.CAPAの7ステップ
シックスシグマと似た品質活動
ステップ1 問題を検出し明確にする
ステップ2 問題を詳しく調査する
ステップ3 根本原因を特定する
ステップ4 是正或いは予防する為の処置をとる
ステップ5 処置された結果を文書化
ステップ6 処置を施した結果を検証する
ステップ7 効果の確認を行う
8.ワークショップ
規制要求と改善活動の本質を盛り込んだ
CAPAフォーマットの作成
9.ワークショップレビュー
規制要求事項が盛り込まれたフォーマット解説
コース概要・お客様の声
セミナーのパンフレットは
FDA GMP(QSR)とISO13485の設計管理の要求事項を比べると、さほどの違いはありませんが、ISOに基づく設計管理では、FDAの査察にまずパスすることはありません。 QSRを理解するためには、QSRを補足説明するFDAのガイダンスを学ぶ必要があり、このガイダンスこそがISOと決定的に違うところと言えます。 このセミナーでは、設計管理を補足説明するガイダンス( Design Control Guidance for Medical Device Manufacturers ) を学びますが、このガイダンスは、GE社が製品開発で導入していたDFSS( Design for Six Sigma )とよく似た開発プロセスで、GEシックスシグマ、ブラックベルトの資格持つクオリス・イノーバが、医療機器開発の原則とその本質を事例を挙げてわかりやすく説明する実践セミナーです。 この設計管理ガイダンスを理解せずにQSRを理解することはできないでしょう。 また、FDA査察官向け査察ガイダンス(QSIT)を引用し、査察官の注目するポイントも解説いたします。 開発部門だけでなく、製品企画、品質保証部門の方にも出席していただきたい価値あるコースです。 IVD(体外診)にも対応した内容になっています。 ( FDAガイダンス邦訳付き )
日 時: 2009年6月9日(火曜)
10:00〜16:00
会 場: 東京都千代田区永田町: 薬業健保会館
講 師: クオリス・イノーバ 代表 木村 浩実
受講料: ¥47,000−( 資料・昼食込み )
¥40,000−( 1社2名様以上、或いは
以前弊社セミナーを受講された方 )
* 社内教育記録としてご活用用頂ける修了書を発行しております。
お申し込みは、セミナー申込み画面からお申し込み下さい。 折り返し、振込先、会場等のご案内を差し上げます。
申込締切:6月4日(木曜)
セミナーの内容
1.設計管理の本質的価値
設計の本質的価値を理解する
Water Fall Process
21CFR Subpart C Design Control
2.重要な品質要素 820.30 (a) 総則
経営スタッフの重要な役割
セクション 820.25 要員の教育
教育の重要性
3.設計及び開発計画 820.30 (b)
製品開発全体を管理するマイルストーン
カスタマーニーズの精度をあげる
カスタマー CTQ
ニーズのエンジニアリンク仕様への変換
コンカレントエンジニアリング
4.設計インプット 820.30 (c)
設計管理で最も重要なフェーズ
曖昧なニーズの基準値への変換
トレーサビリティーマトリックス
設計インプットが製品品質を決める
5.設計アウトプット 820.30 (d)
設計アウトプットを定義し明確にする
DMRとのトレーサビリティー
6.設計審査 820.30 (e)
問題の早期発見、解決が目的
設計審査プラン
設計審査は重要な設計品質ポイント
7.設計検証 820.30 (f)
検証と設計バリデーションの相違
全てがトレーサブルとは
設計検証レポートは開発者の日々の活動記録
8.設計バリデーション 820.30 (g)
ユーザーニーズの精度が成否を決める
設計バリデーションとプロセスバリデーション
バリデーションを実施するタイミング
8.設計移管 820.30 (h)
設計移管の重要な要素
DHFとDMR、DHR
9.設計変更 820.30 (i)
設計変更管理手順
重要な査察ポイント
コース概要・お客様の声
セミナーのパンフレットは
Qualys Innova FDA DC Course 0904 SB0809E.pdf
ISOだけの品質システムを運営し、認証機関の監査に慣れている企業にとって、FDA QSR( Quality System Regulation )とは、単にISOの延長線にあるもの、と簡単に片付けられているようです。 しかし、米国民の健康を守るというミッションを掲げて海外まで査察にやっているFDAと、ISO監査を比べることはそもそも間違っています。 ISO監査員はシステムが維持されているかと問い、FDA査察官はなぜ品質不良が発生するのかと問うのです。 この事を理解するためには、QSRを補足説明しているFDAガイダンスや規制の序文( Preamble )を理解する必要がありますが、本コースでは、FDA査察の際に明確に宣言される4つのエリア、1.マネジメントコントロール 2.設計管理 3.CAPA 4.P&PC をメインに、FDA査察官向け査察ガイダンス(QSIT)、FDA査察解析データを元にFDA規制要求事項の本質をわかりやすく説明します。 また、全世界に100以上の工場を持つGEヘルスケアのコーポレートオーディターとして世界の工場を監査した豊富な実績とFDA査察経験、GEシックスシグマ・ブラックベルトとして多くの品質改善経験をしてきた実績から医療機器開発・製造の本質を鋭く説く実践コースです。
日 時: 2009年5月20日(水曜) 10:00〜16:00
会 場: 東京都千代田区永田町: 薬業健保会館
講 師: クオリス・イノーバ 代表 木村 浩実
受講料: ¥47,000−( 資料・昼食込み )
¥40,000−( 1社2名様以上、或いは以前弊社セミナーを受講された方 )
* 社内教育記録としてご活用用頂ける修了書を発行しております。
お申し込みは、セミナー申込み画面からお申し込み下さい。 折り返し、振込先、会場等のご案内を差し上げます。
お申し込みの受付を終了いたしました。
セミナーの内容
1.FDA規制の本質
FDAのミッション
FDAの組織
米国の法規制
ISOとQSRの本質的な違い
理解すべきガイダンス
情報の入手
2.FDA査察の本質
FDA査察とは
査察の頻度
品質不要が査察リスクを高める
Warning Letter
FDA査察対応準備
FDA査察の本質
3.QSITとは
QSIT査察とは
QSITアプローチとは
FDA解析データ
4つのフォーカスエリア
4.規制要求事項の解説
820.20 経営者の責任
820.22 品質監査
820.25 要員
820.30 設計管理
820.40 文書管理
820.50 購買管理
820.60 識別
820.65 トレーサビリティー
820.70 生産及び工程の管理
820.72 検査・測定及び試験装置
820.75 工程の妥当性確認
820.80 受領、工程内及び完成機器の受入
820.86 受入の状態
820.90 不適合品
820.100 是正処置及び予防措置
820.120 ラベリング
820.130 機器の包装
820.140 取扱
820.150 保管
820.160 流通
820.170 据付
820.180 記録
820.181 DMR
820.184 DHR
820.186 品質システム記録
820.198 苦情ファイル
820.200 付帯サービス
820.250 統計的手法
コース概要・お客様の声
セミナーのパンフレットは
Qualys Innova FDA QSR Course 0904 SB0811d.pdf
医療機器・IVDのためのFDA GMP、21CFR Part 820 QSR ( Quality System Regulation ) サブパートG 「生産及び工程管理」、及び QSR で規制されているP&PCは、FDA査察官向け査察ガイド(QSIT)の4つの査察重要項目の内の一つであり、査察官が注目する項目です。 このコースでは、製造工程の検証方法とプロセスバリデーションの実際の方法を、FDAガイダンス( Guideline on General Principles of Process Validation )のみならず、GHTFガイダンスGHTFガイダンス( Quality Management Systems – Process Validation Guidance )、医薬品GMP、FDA Warning Letter などさまざまな角度から要求事項を紐解き、体外診断薬の工程をメインに例示し( Guideline for the Manufacture of In Vitro Diagnostic Products )、製造工程を科学的根拠、妥当性をもって設計し、検証するプロセスを学びます。 医療機器・体外診断薬製品の開発、製造、品質保証部門の方に最適のコースです。
日 時: 2009年7月14日(火曜)
10:00〜16:00
会 場: 東京都千代田区永田町: 薬業健保会館
講 師: クオリス・イノーバ 代表 木村 浩実
受講料: ¥47,000−( 資料・昼食込み )
¥40,000−( 1社2名様以上、或いは以前弊社セミナーを受講された方 )
* 社内教育記録としてご活用用頂ける修了書を発行しております。
お申し込みは、セミナー申込み画面からお申し込み下さい。 折り返し、振込先、会場等のご案内を差し上げます。
申込締切: 7月9日(木曜)
セミナーの内容
1.製造工程を管理する目的とその本質的価値
設計仕様に適応するということ
なぜ工程バリデーションを実施するのか
2.法的要求事項
21CFR820 Subpart G
QSIT( FDA 査察ガイド )
FDA プロセスバリデーションの一般通則
GHTF プロセスバリデーションガイダンス
IVD 製造業者のためのガイドライン
3.生産及び工程管理 820.70
4.検査・測定及び試験装置 820.72
5.工程の妥当性確認 820.75
6.工程の検証とバリデーション
QC工程表(図)を用いた検証方法
バリデーションされるべき工程
バリデーションの考え方と進め方
バリデーションの種類
バリデーション計画
バリデーションマスタープラン
設計時適格性の確認(DQ)
据付時適格性の確認(IQ)
キャリブレーション
稼働性能適格性の確認(OQ)
製造性能適格性の確認(PQ)
レポート
変更管理と再バリデーション
7.IVD工程に於けるバリデーションの例
工程のマッピング
工程の重要な要素
( 建物・水・空気 )
8.コンピューターバリデーション
バリデーションを行う理由
バリデーションの進め方
コース概要・お客様の声
セミナーのパンフレットは
21CFR Part 820 QSR subpart G 「生産及び工程管理」が要求する工程検証と工程バリデーションについて、FDAプロセスバリデーションの一般通則、GHTFプロセスバリデーションガイダンス、医薬品GMP、FDA Warning Letter などさまざまな角度から要求事項を紐解き、体外診断薬の工程をメインに例示し、製造工程を科学的根拠、妥当性をもって設計し、検証するプロセスを学びます。 医療機器・IVDの開発、製造、品質保証部門の方に最適なコースです。
日 時: 2009年3月11日(水曜) 10:00〜16:00
会 場: 東京都千代田区永田町: 薬業健保会館
講 師: クオリス・イノーバ 代表 木村 浩実
受講料: ¥47,000−( 資料・昼食込み )
¥40,000−( 1社2名様以上、或いは以前弊社セミナーを受講された方 )
お申し込みは、セミナー申込み画面からお申し込み下さい。 折り返し、振込先、会場等のご案内を差し上げます。 お問い合せは、お問い合せページからお願いします。
セミナーの内容
1.製造工程を管理する目的とその本質的価値
設計仕様に適応するということ
なぜ工程バリデーションを実施するのか
2.法的要求事項
21CFR820 Subpart G
QSIT( FDA 査察ガイド )
FDA プロセスバリデーションの一般通則
GHTF プロセスバリデーションガイダンス
IVD 製造業者のためのガイドライン
3.生産及び工程管理 820.70
4.検査・測定及び試験装置 820.72
5.工程の妥当性確認 820.75
6.工程の検証とバリデーション
QC工程表(図)を用いた検証方法
バリデーションされるべき工程
バリデーションの考え方と進め方
バリデーションの種類
バリデーション計画
バリデーションマスタープラン
設計時適格性の確認(DQ)
据付時適格性の確認(IQ)
キャリブレーション
稼働性能適格性の確認(OQ)
製造性能適格性の確認(PQ)
レポート
変更管理と再バリデーション
7.IVD工程に於けるバリデーションの例
工程のマッピング
工程の重要な要素
( 建物・水・空気 )
8.コンピューターバリデーション
バリデーションを行う理由
バリデーションの進め方
セミナーのパンフレットはこちらからダウンロードできます。
FDA Process Validation Course SB0902.pdf
2009年セミナーをご案内いたします。 ( updated: 6 Sep. )
各コースの概要、過去に開催されたセミナーについては、FDAセミナーのページを御覧下さい。 各セミナーへのお申し込み、受講料等の詳細は、約1ヶ月半前にこのページでお知らせいたします。 その後セミナー申込みページからお申し込み頂けます。 セミナーに関するお問い合せはこちらからお願いします。
* 日本でのFDA査察が活発化しておりまして、お客様のFDA査察サポートの為、予定通りセミナーが実施できない場合がございます。 予めご了解下さい。
< 大切なお客様へ >
お客様のFDA査察準備の為、10、11月のセミナーはキャンセルとなりました。 予定をして頂いていたお客様には、大変ご迷惑をお掛け致しますことを心よりお詫び申し上げます。 状況をご理解の上、了解頂けますようお願い申し上げます。 尚、年内中のオンサイトセミナーは11月以降可能ですのでお問い合せ下さい。 ( 2010年のセミナー予定は、年内に発表いたします。 )

FDAの査察で依然として最も多い指摘のカテゴリーがCAPAです。 FDAは、品質を改善するためのCAPA( 是正処置及び予防措置 )を見ること で、規制順守のみならず、企業の品質活動に対する姿勢を確認しようとします。 FDAのCAPAに関する規制要求事項をFDA Warning Letter などから学び、規制要求事項と実際に皆様の会社で使用している是正処置及び予防措置のフォーマットを比べ、FDAの規制に適合するフォーマットを実際に手 を動かして考え、作る実践コースです。 コース終了後には、すぐに会社で仕様できるフォーマットが完成することでしょう。 クオリス・イノーバでは、 100以上の工場を持つGEヘルスケアに於いてコーポレートオーディターとして世界各国の工場を監査し、CAPAで改善させてきた実績と、GEシックスシ グマ、ブラックベルトの資格を持ち、CAPAの7つの改善ステップがシックスシグマに似た品質改善活動である事を豊富な品質改善経験を通して説明します。
コースの詳細とお申し込みはこちらから

FDA GMP:QSR( Quality System Regulation )とISO13485の要求事項を比べてみると、違いはあまりないように思われます。 しかし、査察に耐えられるかとなると、???。 お金を払って認証 される馴れ合いのISO監査と国民を守るために外国までやってくるFDA査察とでは、まったくアプローチが異なります。 QSRを理解するためには、 QSRを補足説明をしている各FDAガイダンスを理解することが欠かせませんが、このコースでは、査察官向けの査察ガイダンス( QSIT )から査察の重要を説明し、FDA規制要求事項の本質を学びます。 全世界に100以上の工場を持つGEヘルスケアのコーポレートオーディターとして世界 の工場を監査し、FDAの査察経験の豊富なクオリス・イノーバならではの他社セミナーにはない実践セミナーです。

私たちが FDA QSR を理解する上で最も重要なセミナーと位置づけているメインコースです。 QSRを理解するためには、ガイダンスの理解が欠かせません。 特に設計管理ガイ ダンスを理解することで、医療機器開発の本質を理解することができ、FDAの規制要求事項の本質が見えてきます。 製品開発のみならず、製品企画、品質保 証部門の方に受講して頂きたい価値あるコースです。 IVD( 体外診 )の方にも受講いただける内容です。
7月14日から2日間、米国コンサルティング会社と共同開催するFDAセミナーです。
修了証書をお渡ししています。
本年2月に東京で開催し、大変好評を頂きました FDA QSR の Basic Course を大阪会場でも実施することになりました。
このコースでは、QSR の要求事項を判りやすく説明しする、実務に大変役立つと好評なコースです。このコースでは要求事項の全てについて、その本質とISO13485との相違 を説明する解説コースです。前回ご出席できなかった方、西日本のの方々が出席できるように致しました。
本年2月に東京で開催した米国コンサルティング会社によるFDAの最新トピックスと、FDA CAPA のワークショップを大阪で開催致します。
このコースでは、ケン氏による FDA のホットな話題をお伝えし、CAPA ワークショップでは、FDA 査察の指摘の大半を占める CAPA をより理解することで、査察に対応し、改善実務に役立つワークショップです。会社に持ち帰り翌日から使えるCAPAフォーマットを作成します。Six Sigma にも通じる CAPA を学ぶことで QSR、CAPA に関する認識が違ってきます。
米国コンサルティング会社が講師を行うコースは通訳付きです。
7月9日から3日間、米国コンサルティング会社と共同開催するFDAセミナーです。
修了証書をお渡ししています。
FDA 査察の準備(午前)及び FDA 査察後、Form 483、Warning Letter、出荷停止、差押えなど一連の FDA の法的アクションとその対処法について説明します。
FDA のホットな話題も含め、FDA 査察の準備についての入門コースです。
QSR Basic Training Course では時間の関係上、規制の説明だけとなりますが、本コースでは要求事項を深く掘り下げ、カスタマーニーズの設計インプットへの展開から検証、妥当性確認、設計トランスファーまで実務に役立つ具体的な例示を上げて説明をする、日本では恐らく開催されることの無い設計管理の実務コースです。
開発担当者はもちろん、品質システム、監査担当の方もご出席して設計の本質と監査のポイントを学ぶことができます。

設計管理コースの様子
通常アメリカで3日間開催されるコースの基礎編を開催いたします。医療機器に特化したソフトウェアバリデーションコースは日本ではまあり開催されていません。FDA が何を要求しているのか理解していただくコースです。
米国コンサルティング会社が講師を行うコースは通訳付きです。
2月14日から2日間、米国コンサルティング会社と共同開催するFDAセミナーです。
星陵会館において、米国コンサルティング会社と共同で FDA GMP ( QSR ) Basic Training Course を開催致します。(1日コース) 定員30名。 早期申込み、2日出席割引がございます。
星陵会館において、米国コンサルティング会社と共同で CAPA Training Course を開催致します。(半日コース)米国FDA最新トピックス(半日コース)も開催、FDA情報に詳しい米国コンサルティング会社から最新の情報を皆様にお伝えします。





FDAセミナーについて
ソリューション
Copyright © Qualys Innova. Co Ltd All Rights Reserved.





