FDA QSR ワンポイントレッスン
FDA QSR の要求事項について、要点をまとめてみました。 皆様からのリクエストにもお答えしてポイントレッスンを進めて参ります。 ご質問は こちらから。
QSR って何?
QSR は Quality System Regulation の略で、米国版医療機器 GMP ( Good Manufacturing Practice ) です。 詳しくは、21 CFR Part 820 が規制の番号になり、CFR とは、Code of Federal Regulation の略で、アメリカ連邦政府諸機関が出した規制集の事を言います。 この規制集には50までのタイトルがあって、医療機器は21番目の Food & Dragに区分けされています。 その中の8巻目に医療機器の規制が載せてあり、820 番目が QSR に相当します。
ISO 13485 との相違は?
改正された薬事法では、グローバルな整合から GMP として ISO 13485 との整合が図られました。 従って、ISO 13485 の認証を取られた企業も多いのではないでしょうか? ( 認証機関から認証を取る法的要求はありません ) FDA も QSR は ISO 13485 と整合していると公に言っています。 では、本当にその言葉を信じていい? FDA のミッションステイトメントは、公衆衛生を守るため、即ちアメリカ国民の健康を守るために、と言う下りから始まります。 ですから、アメリカ国民を守るために、海外にまで出て行って監査ではなく、査察を行うのです。 従って査察の厳しさは、ISO だけをやっていたのでは到底査察に合格できるものではありません。 彼らは不適合の原因や品質不良の原因を統計的に知っていて、そこを査察してきます。 査察官の質問に証拠を見せて回答できるということがポイントになるでしょう。 証拠とは文書であり記録です。
QSR に準拠する心構え
海外に製品を輸出する企業は、薬事法の監査を所轄行政から受け( 国内で販売するときのみ )、ISO13485 或いは ISO9000, 14000 の監査を認証機関から毎年受け、さらに CE マーク、中国に関しては。。と1年中監査、査察だらけ、FDA が査察に来るとなると会社中大騒ぎになります。( 恐らく通常の仕事が3ヶ月できなくなる (*_*) ) おまけに社内で内部監査を毎年行い、全ての監査結果のフォローに明け暮れる。 これはもう品質システムだけの組織がないと対応できないのは明らかですね。 そして会社全体でどれだけの工数がかかっているか計算したことがあるでしょうか? 膨大な時間を監査のために使っていることがおわかりになると思います。 すると、仕方がないからするのか、品質改善やコンプライアンスの一つの手法として積極的に取り組んでいくのかによって、この膨大な工数が生きてくるか、死んでしまうかのどちらかになるかもおわかりでしょう。 考え方一つでコスト削減にもなり得るのです。 品質システムの中でも監査の厳しい FDA QSR は多くのガイダンスが出ています。 QSR の要求事項を深く学ぶことにより、元々品質のよい日本製品は、さらによい品質とコンプライアンスを手に入れることができるでしょう。
品質システムはどう構成すればいい?
ISO 13485 0.4 では、御社の品質システムを FDA の要求事項に合わせて統合してもいいと言っています。 すでに ISO 13485 の品質システムをお持ちであれば、FDA QSR の要求事項に足りない箇所を加えて統合します。 そして、星取り表を作成し、品質システムのどの手順が ISO に適合し、どの手順が FDA QSR に適合しているのか明確にしておけばよいでしょう。 しかし、これは必ず文書化して品質システムに挿入しておかないと、「証拠」を示すことができません。
なんちゃって ISO に気をつける
ISO の認証を取得すると安心し、気分も大きくなってよくこう言われます。 C「 うちは ISO 9000, 13485 の両方取得していますから大丈夫ですよ。」 QI「 それではちょっと見せて頂きますね。」 QI「 ・・あの〜、どうして設計手順も無いのに認証を受けられたんですか (・o・) 」 C「 それは、その・・・ (^_^; 」 QI「 っていうか、そんな認証機関があるんですね・・ (・o・) 」 これは、ある特定の企業さんだけではなく、よくある話で驚いています。 話をよく聞くと、ISO 13485 の人気が高く、監査員が不足していてご自分の得意分野しか詳しく見られない監査員が多いようです。 FDA の査察官は、認証機関 ( Notified Body ) の出す認証書など見向きもしないので注意して下さい。 ( アメリカでは ISO に馴染みがない。 そもそもよその国の規制に関心を示さない。これはもうアメリカの国民性ですね。 ) 断言してもいいですが、ISO 13485 を取得しているからと言って FDA の監査にパスすることはまずあり得ません。 要求事項は同じでも査察官の見る深さが違います。
略語を覚える
査察の際に頻繁に出てくる言葉は、日頃から社内でも使うように工夫しておくことをお勧めします。 査察の際に、(・o・) 状態はまずいですよね。 DHF, DMR, DHR の3つは必ず覚えておきましょう。 DHF ( Design History File ) : 設計管理の項目( 820.30 ) の活動の結果を記録したもの全て。 820.30 で文書化することと記載されている項目はこれに該当すると考えてください。 DMR ( Device Master Record ) : 機器原簿と呼び、820.181 に詳細が説明されています。 薬事法で言う、製品標準書に記載される文書類と考えるとわかりやすいでしょう。 製品を作るのに必要な文書類です。 DHR ( Device History Record ) : 機器履歴簿と呼ばれ、820.184 に詳細説明されています。いわゆる製造記録のことですが、含まれる情報が要求されているので注意して下さい。 ポイントは、社内で規定されている文書リストにどの文書がこれらの文書に相当するかを明記しておくことです。
V & V
V&V は顔文字ではなく、Verification ( 検証 ) and Validation ( 妥当性確認 ) の事です。 よく見かけるのは、検証と妥当性確認を混同、或いは意味を理解しないで使用されているケースです。 この相違は後の設計管理や工程管理のところで説明しますが、ポイントは、規程や手順書の中で明確に定義をしておくことでしょう。
経営者の責任 ( 820.20 )
( 2 ) 意外に見落とされているのが、経営資源の要員の割当です。 例えば、ある工程の作業員の力量が設定されておらず、力量の判定も行っていない作業員が作業をしていることを内部監査で指摘し、報告を受けた経営幹部が何も指示をしていなかったとしたら重大な指摘となるでしょう。 ( 3 ) 管理責任者について、QSR では任命し文書化しろと言っています。 管理責任者の名前、任命責任者のサインの入った文書は、品質システム上で必要です。 ( ii ) 品質システム監査の報告を執行責任者に報告し、レビューを受けているでしょう。 ここまではいいのですが、どういう評価をして報告し、レビュー者のコメント、そのコメントに対する対策が必要です。 ( c ) マネジメントレビューについて、ほとんどの会社では、年に一度しか開催していないのではないでしょうか? それでマネジメントサイドは、会社の品質システム上の問題点を充分な頻度で把握している。。。とは言えないのです。 では、4半期に1度でいいでしょうか?? それでも査察で指摘を受けた会社があるということだけ述べておけば頻度についてはおわかりになるでしょう。
要員 ( 820.25 )
必要な教育・経歴・訓練及び経験を有する充分な要員を雇用し。。と要求されています。 社員に必要な教育を。。 私はどの規程やSOPを知っておくべきなの? この質問に答えられなければ、この要員で指摘されることになるでしょう。 経験的にCAPAを回すと、ほとんどの根本原因が手順を知らない、教育を受けていない、というところに集約されます。 ということは。。。
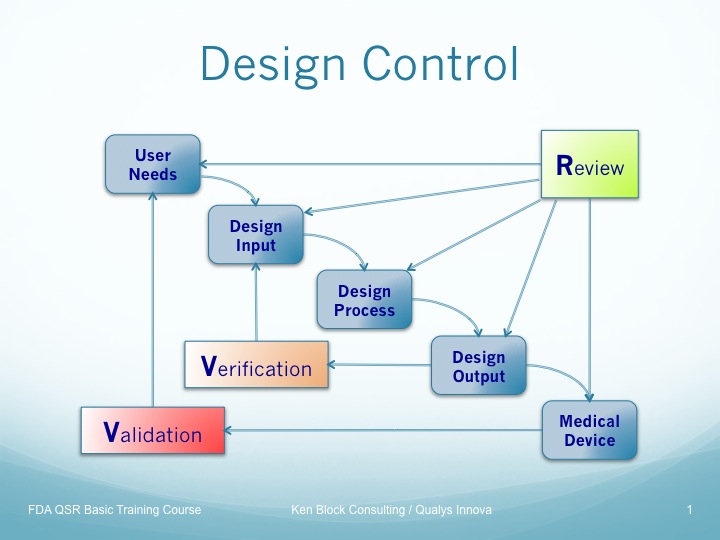
設計管理 ( 820.30 )
安全性、有効性を含む医療機器の品質は設計段階で確立されます。 設計管理の手順では、開発の計画から製造部門への設計移管まで、QSRの要求する項目に従い、手順があるべきです。 FDAは設計ガイドラインで、一つの概念図を利用して設計管理を説明しています。 この図は、開発の各ステップにデザインレビューが必要であること、検証と妥当性確認の関係を明確に示しています。
 HOME
HOME 前のページへ
前のページへ
{kind=link}